





L’attuale definizione di Paralisi Cerebrale Infantile elaborata da Rosenbaum nel 2006, ci parla di “un gruppo di disturbi permanenti dello sviluppo del movimento e della postura, che causano una limitazione delle attività, attribuibili ad un danno permanente (non progressivo) che si è verificato nell’encefalo nel corso dello sviluppo cerebrale del feto, del neonato o del lattante. I disturbi motori della PCI sono spesso accompagnati da disturbi sensitivi, sensoriali, percettivi, cognitivi, comunicativi, comportamentali, da epilessia e da problemi muscoloscheletrici secondari “(Rosenbaum et al.,2006). Poiché in età evolutiva parliamo della crescita come di uno sviluppo armonico dove le aree motorie, sensitive, cognitive, linguistiche, sociali e affettive s’interconnettono tra di loro portando a una progressiva maturazione nei processi di adattamento ambientali, possiamo comprendere di come una lesione cerebrale, in particolar modo nei primi dodici mesi di vita, necessiti di progettualità riabilitativa multidisciplinare. La lesione cerebrale può essere determinata da fattori prenatali a insorgenza in età fetale, fattori perinatali correlati ad un evento che insorge nelle ore o giorni immediatamente successivi al travaglio e fattori postnatali, quando avvengono più tardivamente nel primo anno di vita. Le cause di PCI in epoca prenatale possono ricondursi ad anomalie genetiche, eventi ischemici, malformazioni cerebrali (come ad esempio la microcefalia o l’idrocefalo) e infezioni del complesso T.O.R.C.H. (toxoplasmosi, rosolia, citomegalovirus, herpesvirus). I fattori perinatali (48 ore prima della nascita – 7 giorni dopo la nascita) e postnatali (7 giorni dopo la nascita – 12° mese di vita) rappresentati da problematiche per lo più di tipo emorragico e asfittico, sono legati ad alcuni parametri importanti come il peso alla nascita e l’età gestazionale. A testimonianza di ciò, un insulto asfittico con conseguente ipossia e necrosi dei territori vascolari di confine comporterà nel bambino pretermine un interessamento della sostanza bianca situata nell’angolo esterno dei ventricoli laterali, condizione espressa con il termine di leucomalacia periventricolare, mentre nel bambino nato a termine lo stesso processo fisiopatologico porterà a una lesione più superficiale denominata leucomalacia sottocorticale. Le diverse correlazioni anatomo- funzionali sono determinate dal fatto che nell’ultimo trimestre di gestazione il cervello del feto va incontro a innumerevoli processi di plasticità cerebrale, inclusi cambiamenti nella sua vascolarizzazione. Nel bambino nato pretermine le cause più frequenti di PCI risultano essenzialmente due: la leucomalacia periventricolare e l’emorragia intraventricolare. Quest’ultima consiste nella rottura di vasi sanguigni a livello della matrice germinativa, sede degli spongioblasti che sono i precursori della corteccia cerebrale: la sua ricca irrorazione (soprattutto tra la 22° e la 24° settimana gestazionale) e la difficoltà del sistema vascolare del pretermine nel regolare le variazioni pressorie, la rendono di conseguenza un bersaglio sensibile per lesioni di natura emorragica. Nel nato a termine i processi fisiopatologici esitanti in PCI con maggiore incidenza in letteratura sono l’encefalopatia ipossico-ischemica e l’infarto cerebrale, chiamato più comunemente con il termine inglese di stroke. L’encefalopatia ipossico-ischemica è una reazione patologica che s’instaura conseguentemente ad un insulto asfittico prolungato: come riportato da Cioni e Ferrari “i meccanismi attraverso i quali questo danno si realizza consistono in una combinazione di fattori cerebrali locali, circolatori e metabolici, e sono legati alla durata e alla severità dell’asfissia “(Cioni & Ferrari,2005). La mancanza di ossigeno comporta in prima istanza una ridistribuzione del flusso ematico verso organi vitali: tuttavia il prolungarsi dell’insulto asfittico causa un ipotensione sistemica che determina lesioni ai territori spartiacque arteriosi, localizzati a livello parasagittale. L’infarto cerebrale, patologia più rara da riscontrare nel paziente in età evolutiva, comporta l’ischemia e la necrosi di una porzione di tessuto nervoso per occlusione di un’arteria cerebrale dovuta alla formazione di emboli o trombi all’interno del vaso sanguigno. Nei nati pretermine la forma di PCI più rappresentata è la diplegia spastica, mentre nei nati a termine è l’emiplegia spastica ad avere incidenza maggiore. Le tetraplegie e le forme discinetiche e atassiche hanno percentuali minori nella popolazione in età evolutiva: tuttavia la complessità del quadro clinico da esse derivanti, necessità sia una presa in carico multidisciplinare sia un continuo aggiornamento da parte dei professionisti sulla ricerca di metodiche riabilitative più efficaci e volte al benessere del bambino e del suo nucleo familiare, come espresso dal modello ICF. Abbiamo pertanto introdotto i termini discinetico, atassico e spastico: esse sono le possibili sequele motorie derivanti dalla lesione cerebrale. Per spasticità intendiamo “l’anomalo reclutamento di unità motorie, velocità dipendente in risposta allo stiramento del muscolo “(Cioni & Ferrari,2005) e pertanto ci rivolgiamo ad una condizione nella quale si manifesta una brusca contrazione in un muscolo in relazione al suo stiramento, condizionato dalla velocità in cui ciò avviene durante l’esecuzione di un movimento. La definizione di discinesia è “un aumento della quantità di movimento (ipercinesia) che porta ad un disturbo della coordinazione motoria e regolazione del tono muscolare “(Cioni & Ferrari,2005): quindi il disturbo motorio in questione comporta la produzione movimenti involontari non controllati, i quali si manifestano nei segmenti corporei non direttamente coinvolti in azioni/gesti finalizzati. Infine per atassia intendiamo “un disordine nell’integrazione nel tempo e nello spazio di schemi di movimento per altro normali. Turba della coordinazione e della statica che, indipendentemente dalla debolezza muscolare, altera la direzione e l’ampiezza del movimento e ne compromette la contrazione muscolare volontaria e riflessa necessaria per mantenere la postura e l’equilibrio “(Cioni & Ferrari,2005): a risultare problematica è la coordinazione motoria e l’integrazione di schemi di movimento e delle sue variabili (intensità, ampiezza e velocità) nello spazio e nel tempo. Le sequele motorie non sono le uniche presenti nelle PCI: ad esempio le forme diplegiche cado-cado e tirati su, individuate da Cioni e Ferrari nella loro classificazione, si distinguono per una componente percettiva più importante e caratteristica rispetto al deficit motorio. Negli esempi menzionati il bambino cado-cado ha degli elementi dispercettivi legati alla ridotta tolleranza dello spazio posteriore, con vertigini e costante sensazione di cadere nel vuoto; al contrario la caratteristica principale del bambino tirati su è rappresentata dalla soppressione periferica delle informazioni cinestesiche (Cioni & Ferrari,2005), ovvero dal trascurare le informazioni propriocettive derivanti dall’ apparato locomotore necessarie per organizzare reazioni di equilibrio e pertanto garanti del mantenimento posturale. Come dimostrato dalle neuroimmagini (RMN, TC ed EG) è stato possibile attribuire delle correlazioni anatomo funzionali tra la sede della lesione e la conseguente tipologia di deficit motorio che da essa ne scaturisce. L’interessamento della sostanza bianca periventricolare, sottocorticale (ad esempio nelle tetraplegie), della capsula interna (struttura contenente fasci di fibre sensitive ascendenti e motorie discendenti) e della corteccia cerebrale è più comunemente associato ad una problematica motoria spastica; il coinvolgimento dei gangli della base, distribuiti sia livello telencefalico sia diencefalico e mesencefalico, comporta una sequela di tipo discinetico; infine la sintomatologia atassica è associata a danni a livello cerebellare. Le anomalie motorie specifiche sono alla base della più comune classificazione delle Paralisi Cerebrali Infantile, di cui parleremo nel paragrafo seguente.
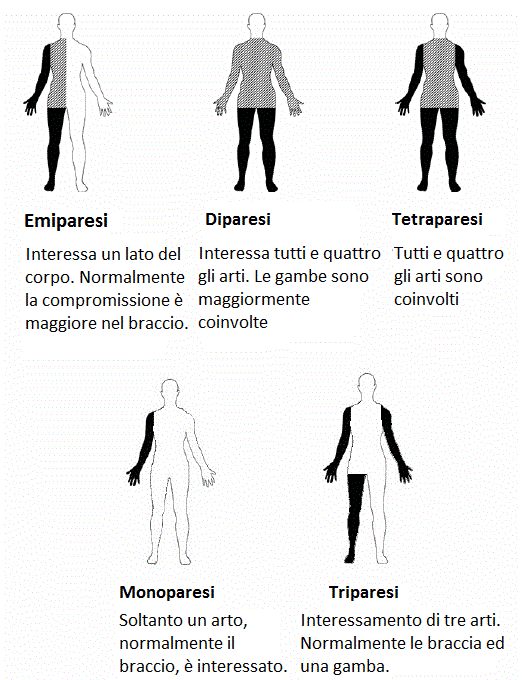 La Paralisi Cerebrale Infantile è stata a lungo oggetto di modifiche nella sua classificazione per via della difficoltà nell’individuare un esatto processo fisiopatologico alla base e per la variabilità del quadro clinico da esso scaturente. La classificazione più comunemente nota è di Hagberg del 1975 il quale all’epoca distingueva la PCI in base sia alla tipologia di deficit motorio prevalente (spasticità, discinesia o atassia) sia alla sua localizzazione topografica (emiplegia, diplegia o tetraplegia) (Hagberg,1975). Le forme cliniche appurate erano pertanto l’emiplegia spastica, la diplegia spatica e la tetraplegia spastica nell’area delle PCI spastiche, le forme coreoatetosiche e distoniche nell’ambito delle PCI discinetiche ed infine nell’ambito delle PCI atassiche la diplegia atassica e l’atassia congenita semplice. Tale classificazione è in realtà molto usata nei giorni nostri perché consente a quadro clinico definito di suddividere sistematicamente i pazienti in categorie ben delineate offrendo vantaggi per scopi epidemiologici, pur tuttavia presentando alcuni limiti: essa non tiene conto sia dei fattori precoci di outcome (poiché non ne stabilisce il processo fisiopatologico ma solo la sequela motoria dominante), sia dell’organizzazione percettivo-motoria dei soggetti patologici nei processi di adattamento ambientale. A tal proposto pare opportuno menzionare altre due tipi di classificazione molto importanti: una è il GMFCS mentre l’altra è la classificazione proposta da Cioni e Ferrari. Il GMFCS, acronimo di Gross Motor Functional Classification System è un sistema di classificazione che tiene conto dell’autonomia grossomotoria raggiunta dal paziente in età evolutiva con diagnosi di PCI. Esso è suddiviso in cinque fasce di età (0-2 anni, 2-4 anni, 4-6 anni, 6-12 anni, 12-18 anni) ed in cinque livelli cui corrispondono gradi decrescenti di autonomia:
La Paralisi Cerebrale Infantile è stata a lungo oggetto di modifiche nella sua classificazione per via della difficoltà nell’individuare un esatto processo fisiopatologico alla base e per la variabilità del quadro clinico da esso scaturente. La classificazione più comunemente nota è di Hagberg del 1975 il quale all’epoca distingueva la PCI in base sia alla tipologia di deficit motorio prevalente (spasticità, discinesia o atassia) sia alla sua localizzazione topografica (emiplegia, diplegia o tetraplegia) (Hagberg,1975). Le forme cliniche appurate erano pertanto l’emiplegia spastica, la diplegia spatica e la tetraplegia spastica nell’area delle PCI spastiche, le forme coreoatetosiche e distoniche nell’ambito delle PCI discinetiche ed infine nell’ambito delle PCI atassiche la diplegia atassica e l’atassia congenita semplice. Tale classificazione è in realtà molto usata nei giorni nostri perché consente a quadro clinico definito di suddividere sistematicamente i pazienti in categorie ben delineate offrendo vantaggi per scopi epidemiologici, pur tuttavia presentando alcuni limiti: essa non tiene conto sia dei fattori precoci di outcome (poiché non ne stabilisce il processo fisiopatologico ma solo la sequela motoria dominante), sia dell’organizzazione percettivo-motoria dei soggetti patologici nei processi di adattamento ambientale. A tal proposto pare opportuno menzionare altre due tipi di classificazione molto importanti: una è il GMFCS mentre l’altra è la classificazione proposta da Cioni e Ferrari. Il GMFCS, acronimo di Gross Motor Functional Classification System è un sistema di classificazione che tiene conto dell’autonomia grossomotoria raggiunta dal paziente in età evolutiva con diagnosi di PCI. Esso è suddiviso in cinque fasce di età (0-2 anni, 2-4 anni, 4-6 anni, 6-12 anni, 12-18 anni) ed in cinque livelli cui corrispondono gradi decrescenti di autonomia:
La classificazione di Cioni e Ferrari mette al centro l’organizzazione funzionale del paziente con PCI, individuando le strategie percettivo-motorie messe da lui in atto per risolvere le problematiche ambientali: all’interno delle categorie proposte da Hagberg è infatti possibile distinguere bambini con una medesima nomenclatura patologica (es. Tetraparesi spastica) ma che organizzano la loro motricità in modo più o meno adattivo confrontati con altri pazienti della stessa categoria. Secondo Cioni e Ferrari le tetraparesi vengono pertanto suddivise in tetraplegie aposturali, acinetiche (caratterizzate da difesa antigravitaria in flessione), a tronco orizzontale ed a tronco verticale, con quest’ultime che riusciranno a raggiungere la statica eretta ed uno schema primitivo deambulatorio con l’utilizzo di facilitazioni (Cioni & Ferrari,2005). Le diplegie spastiche vengono suddivise in base ad un deficit prevalentemente percettivo o al contrario prettamente motorio nelle quali la deambulazione rappresenta il fattore discriminante. I pazienti diplegici in età evolutiva potranno organizzare un cammino propulsivo, a gonna stretta, a funambolo o a temerario (Cioni & Ferrari., 2005). Le emiplegie spastiche presentano una classificazione che combina l’organizzazione funzionale con il tipo/timing di lesione alla base: si distingueranno pertanto le forme malformative precoci, prenatali, connatali (o perinatali) e acquisite (Cioni & Ferrari,2005). In conclusione non è ancora disponibile, nonostante il potenziamento di strumenti di neuroimmaging, una classificazione che raggruppi al tempo stesso bambini con PCI sia in base al processo fisiopatologico (a parte il caso unico dell’emiplegia spastica), sia in base al loro orientamento prognostico. In ambito clinico lo strumento tuttora più noto e di facile utilizzo è quello proposto da Hagberg, anche se il modello coniato da Cioni e Ferrari risulta essere più funzionale e pertanto qualitativamente migliore per elaborare un progetto riabilitativo adeguato per il paziente.
Riprendendo quanto scritto nel paragrafo 1.1, la spasticità è “un anomalo reclutamento di unità motorie, velocità dipendente, in relazione allo stiramento del muscolo “(Cioni & Ferrari,2005). Essa è determinata da un danno sia al primo motoneurone localizzato nella corteccia cerebrale che alla sostanza bianca contenente le fibre discendenti motorie. La suddetta lesione comporta l’insorgenza di contrazioni brusche ed improvvise velocità-dipendenti: in seguito a questa contrazione esagerata, il muscolo riscontrerà successivamente difficoltà a rilasciarsi. La spasticità costituisce un difetto sia di natura centrale o top-down come danno alle vie piramidali sia periferica o bottom-up, in quanto costringendo il muscolo in tensioni obbligate prima della normale soglia di stiramento, condiziona l’informazione periferica derivante dai fusi neuromuscolari (Cioni & Ferrari,2005). Nel suo complesso la spasticità comporta una molteplicità di segni clinici quali: alterazioni del tono, della produzione di schemi motori, di scansione temporale della contrazione, difetti di passività (l’abnorme reazione allo stiramento), facile esauribilità e precoce affaticamento. Questo processo determina un progressivo accorciamento muscolare che, se non trattato opportunamente evolve dapprima in contrattura, con il rilascio del muscolo che diventa possibile solo in narcosi ed infine in retrazione, dove è necessario intervenire chirurgicamente a livello di apparato locomotore. La spasticità influisce primariamente nei moduli motori, alcuni dei quali saranno possibili solo in sinergie obbligate (ad es., dorsiflessione della tibio-tarsica solo in condizioni di triplice flessione, oppure flessione del polso ad avambraccio pronato e gomito flesso nell’arto superiore). Più avanti nello sviluppo, i bambini con diagnosi di PCI spastica avranno difficoltà nell’acquisizione di tappe psicomotorie fondamentali come il raggiungimento della posizione seduta, della statica eretta e della deambulazione: tali competenze non saranno possibili in tutti i soggetti poiché in alcuni di essi, l’entità della lesione neurologica impedirà l’instaurarsi di basilari abilità antigravitare.
Le tetraplegie spastiche sono forme di PCI ove si riscontra un interessamento equivalente dei quattro arti e disturbi visivi, uditivi, cognitivi, orofacciali e comiziali molto frequentemente associati. L’organizzazione posturale di questi pazienti rappresenta secondo Cioni e Ferrari il fattore discriminante per categorizzare le sotto forme di tetraparesi spastica: individueremo pertanto tetraplegie aposturali, con difesa antigravitaria in flessione, a tronco orizzontale e a tronco verticale. Per postura intendiamo il mantenimento di una posizione del corpo nello spazio, rispetto ad un sistema geocentrico che ha come riferimenti la forza di gravità ed il piano orizzontale tangente alla superficie terrestre: per far ciò il bambino svilupperà delle competenze antigravitare adeguate sia sulla metà superiore del corpo che su quella inferiore. Nello sviluppo posturo-motorio è fondamentale l’apprendimento delle reazioni di raddrizzamento e di sostegno. Le reazioni di raddrizzamento sono tutti quei movimenti automatici, innescati da informazioni di tipo vestibolare, visivo e propriocettivo che permettono il riallineamento dei segmenti corporei quali capo, tronco e arti nello spazio rispetto all’asse longitudinale: ad esempio raddrizzare il tronco in statica eretta significa portarlo perpendicolarmente al terreno. La reazione di sostegno definisce la capacità degli arti di sostenere il peso del corpo, vincendo la forza di gravità: essa può subire alterazioni di tipo qualitativo o di tipo quantitativo (ad es., ipoposturalità). Nelle tetraplegie spastiche aposturali è impossibile osservare una qualsiasi abilità antigravitaria: questi bambini giacciono al suolo in un atteggiamento definito “a batrace “, con gli arti superiori estesi ed abbandonati ai lati del tronco. In essi l’ipotono può, con il passare degli anni, diventare in seguito ipertono: ciò determina un cambiamento nell’organizzazione della condotta antigravitaria ma non del funzionamento adattivo. Tali pazienti svilupperanno una difesa antigravitaria con i quattro arti in flessione, schema tipico del neonato che non è ancora in grado di organizzare né una reazione di raddrizzamento, né di sostegno. I pazienti con tetraparesi a tronco orizzontale riescono ad organizzare delle reazioni di sostegno sugli arti superiori da prono, estendendo i gomiti, flettendo i polsi ed utilizzando le mani primariamente come appoggio. L’organizzazione di un pattern locomotorio ed il mantenimento della statica eretta costituiscono due tipologie di abilità motorie proibitive in questi bambini. Secondo Ferrari esiste una variante di questa forma rappresentata dalla tetraplegia con automatismi sottocorticali, in grado di evocare un schema primitivo deambulatorio grazie al sostegno posteriore da parte dell’adulto: in realtà tali pazienti avanzano propulsivamente, con il tronco retropulso rispetto agli arti inferiori e con questi ultimi non in grado di accettare il carico nel ciclo del passo. Le tetraparesi con maggiori capacità motorie sono quelle con antigravità bipedica o a tronco verticale. In esse sono presenti competenze antigravitarie sia sugli arti superiori con uno schema flessorio in afferramento, sia sugli arti inferiori che al contrario presentano una schema globalmente estensorio nonostante la componente flesso-adduttoria delle anche e la facile esauribilità della reazione di sostegno. Questi pazienti riescono a raddrizzare il tronco in statica eretta grazie alla fissazione distale, stabilizzando l’asse corporeo in senso centripeto. Il cammino in questi bambini si presenta lento e faticoso per via delle difficoltà nell’integrare la reazione di sostegno con la reazione segna passi, cioè con il programma motorio che presiede al movimento alternato degli arti inferiori con sede nel midollo spinale. Anche se esercitata nelle sedute di terapia, in età adolescenziale la funzione cammino verrà comunque abbandonata per orientare il paziente verso l’utilizzo di una carrozzina ortopedica: il motivo di tale scelta è da ricondursi al progredire delle deformità muscolo- scheletriche ed una maggior funzionalità del suddetto ausilio nei confronti delle esigenze ambientali dell’individuo. Secondo dati aggiornati al 2012, l’incidenza della tetraplegie spastiche è del 7 % di tutte le PCI (Fabbro et al.,2012) e il fattore causale primario è rappresentato dalla leucomalacia periventricolare (Cioni & Ferrari,2005).
Per diplegie spastiche intendiamo tutte quelle forme di Paralisi Cerebrale nella quale la lesione comporta un interessamento prevalente ma non esclusivo di due arti simmetrici, più comunemente quelli inferiori. Gli AAII (arti inferiori) sono contraddistinti da iperriflessia, aumento del tono e limitazioni nei moduli motori; al contrario gli AASS (arti superiori) presentano buone abilità grossomotorie con difficolta che persistono nell’ambito della motricità fine legate in gran parte all’integrazione percettivo-motoria. Questi pazienti riescono a raggiungere, nel corso del loro sviluppo, tappe psicomotorie fondamentali come l’acquisizione della statica eretta e della funzione cammino entro i 5 anni: il cammino verrà mantenuto per un periodo variabile della loro vita in relazione all’instaurarsi di deformità muscolo-scheletriche. Secondo la classificazione di Cioni e Ferrari, il vero fattore discriminante delle diplegie spastiche è rappresentato dalle diverse strategie messe in atto nella funzione cammino. In base a tale prospettiva i pazienti con diplegia spastica sono stati raggruppati nelle sotto forme propulsivi, a gonna stretta, funamboli e temerari. Nella forma diplegica propulsiva, la quale si può ulteriormente categorizzare a seconda dell’atteggiamento flessorio o estensorio dell’anca in appoggio monopodalico, gli elementi connotativi sono rappresentati dell’antepulsione del tronco e dal bilanciamento sulle punte. La propulsività si traduce nel costante inseguimento del baricentro da parte del paziente, proiettato nell’emispazio anteriore dal tronco antepulso e dal bacino antiverso: il carattere propulsivo potrebbe derivare dalla difficoltà nel bambino di organizzare reazioni paracadute efficaci nell’emispazio posteriore (Cioni & Ferrari,2005).
Nei pazienti con diplegia a gonna stretta il cammino si presenta con un’accentuazione della flessione di ginocchio nell’arto in carico, con intrarotazione e limitazione nel range di movimento dell’anca sia nella sua flessione(in fase di oscillazione) che nella sua estensione (nella fase di appoggio) e con bilanciamento sull’avampiede: il tronco tuttavia si mantiene verticale determinando buone abilità di fissazioni prossimali e di conseguenza un cammino qualitativamente migliore rispetto alla prima forma. Un segno clinico peculiare delle diplegie a gonna stretta è il pendolo di bacino sul piano sagittale per consentire l’avanzamento nel passo anteriore. Questi bambini incorrono precocemente in retrazioni muscolo-scheletriche con frequente coinvolgimento di ischiocrurali, adduttori della coscia e tendine sottorotuleo: in questi casi diventa inevitabile il ricorso alla chirurgia funzionale per preservare la deambulazione, fin quando risulta possibile. I diplegici della forma funamboli presentano problematiche dispercettive più importanti come la ridotta tolleranza dello spazio posteriore. Il cammino è definito a funambolo perché essi effettuano un particolare pendolo di tronco che ricorda quello vagamente utilizzato dagli equilibristi: altri elementi distintivi sono il pivot sull’avampiede dell’arto in appoggio (equinismo di spinta), arti superiori abdotti e semiflessi ai gomiti, l’antepulsione del tronco e la velocizzazione della marcia. All’interno del gruppo dei diplegici definiti come temerari, sono incluse altre tre sottoforme che si differenziano in base alla localizzazione del deficit motorio: distingueremo pertanto una sottoforma generalizzata, una distale propriamente detta e una asimmetrica, comunemente chiamata con il termine di doppia emiplegia. Nei temerari generalizzati, gli elementi connotativi della funzione cammino sono l’equinismo all’avvio della marcia e la modesta flesso-adduzione delle anche. Nei temerari a carattere prettamente distale a fronte di un minor interessamento dell’anca, l’equinismo è più marcato e risulta osservabile in fase di pieno appoggio, fase di oscillazione e nell’iniziale contatto. Per quanto concerne le doppie emiplegie, esse presentano un emilato più colpito nella quale anca e ginocchio rimangono flessi in fase di appoggio monopodalico. Il deficit motorio è minore rispetto alle altre due sotto forme: ciò nonostante, il più ampio coinvolgimento dell’arto superiore nell’emilato peggiore, specie in attività motorie complesse come la corsa, rendono questi pazienti più simili ai veri emiplegici (Cioni &Ferrari,2005). A fronte di problematiche motorie minori rispetto alle tetraplegie, le diplegie spastiche presentano tuttavia importanti disturbi sia percettivi, come la ridotta tolleranza del vuoto circostante, sia piscopatologici, poiché soggetti dotati di un funzionamento cognitivo qualitativamente migliore e di conseguenza più consapevoli delle loro difficolta. Le diplegie spastiche sono patologie molto diffuse in età evolutiva in quanto si ritrovano nel 45 % della totalità dei casi con PCI (Fabbro et al.,2012).
Le emiplegie spastiche sono patologie derivanti da una lesione unilaterale o asimmetrica a livello del SNC che si manifesta nel paziente con deficit motori e di altra natura, maggiori (o esclusivi) nella metà di corpo controlaterale rispetto al danno cerebrale e con l’interessamento sia dell’arto superiore che di quello inferiore. Difetti percettivi e disturbi delle funzioni corticali superiori come ad esempio la negligenza spaziale unilaterale e l’epilessia, gravano sul quadro clinico complicando l’instaurarsi di funzioni adattive perlopiù manipolatorie nel lato plegico. L’integrazione tra i due emilati rappresenta quindi il punto centrale dell’approccio riabilitativo. Le sotto forme di PCI che tratteremo saranno pertanto: le forme malformative precoci, le forme prenatali, le forme connatali o perinatali e le forme acquisite (Cioni et al.,1999). Nelle forme malformative precoci la lesione avviene tra il primo ed il secondo trimestre di gravidanza: disturbi di natura vascolare, infettiva e legati alla proliferazione ed alla migrazione neuronale sono da considerarsi alla base del danno (Cioni & Ferrari,2005). Il pattern motorio scaturente non presenta particolari alterazioni all’ arto inferiore plegico se non tendenza al recurvato di ginocchio in fase di appoggio monopodalico ed equinismo di sospensione: le problematiche appena elencate non comportano difficoltà nel raggiungimento delle tappe psicomotorie. L’arto superiore dell’emilato plegico presenta condotte patologiche osservabili nella intrarotazione del braccio, nella flessione del gomito, nella pronazione dell’avambraccio, nella flessione del polso e nell’atteggiamento della mano a ventaglio, ovvero con flessione della falangi prossimali nelle dita: i segni appena elencati vengono accentuati nelle reazioni associate che il bambino involontariamente produce durante l’esecuzione di attività motorie complesse. I bambini emiplegici della forma malformativa precoce riescono tuttavia ad apprendere funzioni prassiche adattive come fare una collana o utilizzare le forbici. Le cause dell’emiplegia prenatale sono da ricercarsi in lesioni ipossico-ischemiche che avvengono nel terzo trimestre di gestazione e che sono spesso bilaterali (Cioni & Ferrari,2005). Il quadro clinico che ne emerge è di un interessamento prevalente dell’arto inferiore piuttosto che del superiore nell’emilato plegico: l’anca è addotta e non risolve la flessione in fase di appoggio monopodalico, mentre il piede ha un atteggiamento in equino varo-supinazione. Con l’arto superiore plegico è possibile l’acquisizione della pinza superiore ed i movimenti di prono-supinazione, testimonianza di una funzione manipolatoria qualitativamente migliore rispetto alla prima forma. Le lesioni che scaturiscono quadri di emiplegie connatali sono di natura anossico-ischemica e di tipo infartuale: questi processi patologici causano lesioni molto spesso bilaterali sia a livello telencefalico che diencefalico (Cioni &Ferrari,2005). Nell’arto inferiore plegico le componenti che più risaltano sono la flessione di ginocchio e l’atteggiamento in equino- valgo pronazione del piede, mentre in quello superiore la flessione e deviazione ulnare del polso e la chiusura della mano a pugno con il pollice che viene spesso imprigionato. Nelle attività manipolatorie questi bambini hanno grandi difficoltà ad integrare la mano plegica negli schemi d’azione e più spesso vengono utilizzati distretti più prossimali come polso ed avambraccio per compiti ausiliari. Lesioni di natura vascolare, infettiva, tumorale o traumatica in epoca neonatale (convenzionalmente entro l’anno di vita) sono le cause principali dell’emiplegia acquisita o quarta forma (Cioni & Ferrari,2005). L’arto inferiore, pur utilizzando schemi rigidi di compenso durante l’avanzamento (come l’andatura steppante con accentuazione della flessione di ginocchio ed anca per sopperire ad un piede cadente) è meno colpito dell’arto superiore omolaterale: in quest’ultimo, oltre alle problematiche legate a moduli e combinazioni motorie sono presenti importanti componenti dispercettive di rappresentazione corticale della mano plegica che ne comporta l’inutilizzo in attività bimanuali. Le emiplegie congenite secondo dati aggiornati al 2012 sono le forme più frequenti di PCI nei nati a termine (44 %) e la seconda nei pretermine dopo la diplegia spastica (20%), per un totale di un 35% delle PCI in generale.
Le Paralisi Cerebrali Infantili di tipo discinetico sono caratterizzate dalla presenza di movimenti involontari ed incontrollati di segmenti corporei non direttamente coinvolti in un’azione finalizzata: ciò rende difficoltoso l’esecuzione del gesto ed il mantenimento posturale. La presenza di discinesie è secondo la letteratura correlata ad un danno dei gangli della base, con il primario coinvolgimento del corpo striato, del globuspallidus e del putamen (Fabbro et al.,2012). I processi eziopatologici alla base della lesione sembrano essere rappresentati dal kernittero, sindrome neurologica che vede il depositarsi della bilirubina nei gangli della base e dall’encefalopatia ipossico-ischemica conseguente ad asfissia perinatale: la PCI di tipo discinetico colpisce nella maggior parte dei casi bambini nati a termine. Esse rappresentano il 10 % di tutte le PCI (Fabbro et al.,2012), nonostante dagli anni ’50 si sia ridotta l’incidenza del kernittero. Le tipologie di movimento discinetico citate in letteratura sono la corea, l’atetosi e le distonie. Per corea o movimenti involontari di tipo coreico intendiamo “movimenti rapidi a scatto, a partenza prossimale, né ritmici né ripetitivi”: essi disturbano il mantenimento posturale, costringendo il bambino ad adottare strategie di compenso. I movimenti involontari di tipo atetosico sono più comunemente descritti come “movimenti lenti, torsionali e vermicolari, a partenza ed interessamento distale”. Le distonie vengono invece definite come “movimenti lenti e torsionali che interessano capo, tronco, cingolo pelvico, cingolo scapolare e gli arti sia superiori che inferiori”: essi sono generati da contrazioni sostenute e durature che portano all’instaurarsi di veri e propri schemi distonici. Storicamente nelle PCI di tipo discinetico vengono distinte le forme con manifestazioni coreo-atetosiche da quelle prettamente distoniche. Le prime rappresentano il 30 % di tutte le paralisi cerebrali di tipo discinetico: questi bambini, nonostante il deficit motorio, riescono a raggiungere la stazione eretta ed uno schema deambulatorio. Sono presenti difficoltà anche nella manipolazione (Fabbro et al.,2012). Il restante 70 % è costituito dalle forme di tipo distonico: al contrario delle precedenti, solo ¼ di questi soggetti riuscirà a conquistare la posizione seduta autonoma (Fabbro et al.,2012). Nonostante la classificazione, alcuni segni clinici come l’incapacità ad inibire il movimento, l’instabilità dell’errore, la difficoltà a mantenere una postura simmetrica, disturbi percettivi come l’avoiding di sguardo e tattile e lo sviluppo tardivo ed atipico delle reazioni di raddrizzamento in senso caudo-craniale, si ritrovano sia nella forma coreico- atetosica che in quella distonica. A discapito delle loro notevoli difficoltà motorie, i bambini con PCI discinetica sono in grado di mettere in atto compensi, nel range delle loro possibilità, per eseguire un’azione finalizzata ad uno scopo: ad esempio l’avoiding di sguardo può essere interpretato sia come un segno, sia come un compenso quando il bambino riesce a dissociare temporalmente informazioni tattili e visive in modo funzionale. Altre strategie messe in atto da questi pazienti osservabili durante le attività manipolatorie sono la fissazione distale degli arti con conseguente riduzione del loro grado di libertà, l’approssimazione del gesto al tronco e la produzione di movimenti servo-motori: in particolar modo quest’ultimi sono dei movimenti parassiti volontari ed organizzati (al contrario delle discinesie) che vengono scaricati nella mano non attiva per liberare quella controlaterale, direttamente coinvolta nell’esecuzione del gesto finalizzato.
L’atassia è un deficit motorio caratterizzato da difficoltà nell’integrazione di tempo e spazio in schemi di movimento normali. La lesione è comunemente localizzata a livello cerebellare e poiché il cervelletto risulta coinvolto in una poliedricità di funzioni, non solo motorie, in questi casi si ricorre spesso all’utilizzo del termine “sindrome atassica”. Connesse alle sequele di tipo motorio, rappresentate primariamente sia dal difetto di coordinazione dei gruppi muscolari noto come asinergia che dalla difficoltà nell’elaborare adeguate reazioni di equilibrio, emergono problematiche di tipo visivo (nistagmo cerebellare), linguistico (linguaggio tipico dell’atassico), comiziale, emozionale e cognitivo. Dal punto di vista clinico, questi bambini si caratterizzano nel primo anno di vita per una spiccata ipotonia e per un ritardo dello sviluppo motorio: in particolare la deambulazione viene acquisita solo tra i 4 ed i 10 anni (Fabbro et al.,2012). La marcia atassica presenta degli elementi distintivi come la base allargata per gestire la proiezione del baricentro, le ginocchia iperestese, lo slancio degli arti inferiori durante l’avanzamento e l’esagerazione delle reazioni di equilibrio. Nelle attività manipolatorie questi bambini riscontrano problemi nel calibrare la giusta ampiezza (ipo- ipermetria) e mantenere la traiettoria (dismetria) del gesto: inoltre, la presenza di tremori intenzionali condiziona la buona riuscita dell’atto motorio. Tuttavia anche per quanto concerne la manipolazione, questi bambini sono in grado di organizzare dei compensi efficaci come la distalizzazione del punto fisso in attività di motricità fine e l’approssimazione del gesto all’asse corporeo. Contrariamente a quanto proposto nella classificazione di Hagberg (1975) nella quale tali forme venivano suddivise in atassia congenita semplice e diplegia atassica, Fabbro propone una variante individuando tre diverse forme cliniche in relazione a fattori eziopatologici: le atassie pure, le sindromi atassiche e le atassie acquisite (Fabbro et al.,2012). Le atassie pure sono correlate ad ipoplasia del cervelletto, le sindromi atassiche vengono descritte come “quadri clinici di atassia congenita non evolutiva nei quali i deficit cerebellari sono i sintomi prevalenti “richiamando patologie di natura genetica come ad esempio la sindrome di Joubert mentre le atassie acquisite non progressive sono dovute perlopiù ad infezioni del complesso T.O.R.C.H. in epoca prenatale (Fabbro et al., 2012).
Le patologie neuromuscolari sono un gruppo eterogeneo di malattie su base genetica che coinvolgono specificamente l’unità motoria, costituita dal tratto motoneurone-fibra muscolare. Esse si suddividono in base alla localizzazione del processo eziologico in patologie con un principale interessamento delle fibre muscolari (es. distrofie di Becker e di Duchenne), della placca motrice (es. miastenie), dei nervi effettori (es. neuropatie) e dei motoneuroni delle corna anteriori del midollo spinale (es. SMA). Nelle malattie neuromuscolari, la progressione del deficit neuromotorio determina delle complicanze di natura sistemica che inficiano sulla qualità di vita dell’individuo, rendendo obbligatorio un approccio clinico multidisciplinare: per esempio, nella Distrofia di Duchenne la progressiva degenerazione della struttura muscolare scheletrica comporterà complicanze sul piano cardiaco (es. Cardiomiopatia dilatativa), sul piano respiratorio e sul piano strettamente ortopedico (es. scoliosi). I segni clinici più comuni e precoci di questo gruppo di patologie sono l’ipotonia e l’ipostenia. La valutazione clinica si articola sia in un esame obiettivo che in una routine di esami di laboratorio comprendente dosaggio enzimatico delle CPK, elettromiografia, biopsia muscolare e indagine genetica. Le patologie neuromuscolari ad incidenza più alta nella popolazione sono le distrofie, malattie degenerative intrinseche della muscolatura. La forma di distrofia più comune in età evolutiva è la distrofia di Duchenne (incidenza 1/3.500 maschi nati vivi) caratterizzata da un‘alterazione del gene xp21 sul cromosoma x che comporta una totale assenza di distrofina, proteina del complesso proteico muscolare che svolge un ruolo fondamentale di rinforzo meccanico a livello del sarcolemma (Ratel et al.,2006). Tale patologia, trasmessa da madri portatrici sane a figli maschi o causata dalla mutazione in de-novo del gene xp21, determina dapprima l’instaurarsi di un processo infiammatorio nelle fibre muscolari ed in seconda istanza la loro sostituzione in tessuto connettivo all’interno del sincizio, fenomeno correlato con la minor potenza generata dalla contrazione muscolare. La degenerazione procede secondo una duplice direzione sia in senso prossimo-distale che caudo- craniale: ciò comporterà un iniziale interessamento della muscolatura del cingolo pelvico. L’ipostenia contribuirà in questi pazienti sia ad un ritardo nell’acquisizione che alla successiva difficoltà d’esercizio di attività grossomotorie quali deambulazione, salire e scendere le scale e passaggi posturali: in particolar modo il segno di Gowers, dovuto alla trazione di entrambi gli arti superiori nella transizione supino - statica eretta è considerato un precoce indice prognostico. Il pattern del cammino dei soggetti con distrofia di Duchenne presenta alcuni tratti peculiari come l’andatura anserina, la pseudo ipertrofia del tricipite surale e la lordosi lombare. La strategia che tali pazienti utilizzano per ottimizzare il dispendio energetico nel cammino è costituita dal massimo sfruttamento delle tensioni legamentose al fine di avvicinare i centri articolari alla GRF, come ad esempio nel recurvato di ginocchio in fase di appoggio. Con il progredire della patologia e la comparsa di retrazioni, questi pazienti perderanno l’uso del cammino (prima dei 13 anni) ed andranno incontro a problematiche secondarie: in particolare l’instaurarsi della scoliosi comporta una complicazione del quadro clinico sul piano cardiaco e respiratorio. La distrofia di Becker (1/18.000-1/35.000 maschi nati vivi) è anch’essa determinata da un’alterazione del gene codificante la distrofina, la quale tuttavia risulta presente nel tessuto muscolare a concentrazioni ridotte. In funzione di ciò il processo degenerativo sarà più lento e la funzione deambulatoria verrà conservata più a lungo (in alcuni casi anche oltre i 18 anni), anche se permarranno complicanze secondarie come le anomalie cardiache, principali cause di morte in tali soggetti. All’interno del gruppo delle distrofie è opportuno citare le distrofie miotoniche, patologie ad alta incidenza (1/8.000 nati vivi) che vedono alla base della degenerazione una mutazione genetica autosomica dominante. Le distrofie miotoniche possono essere di tipo DM1 (Distrofia Miotonica di Steinert) o DM2 (forma più rara). Mentre nella DM1 la mutazione del gene codificante per l’enzima miotonina protein chinasi (DMPK) comporta, oltre a danni sistemici (ad esempio a livello cardiaco, cerebrale, endocrino e nel corpo vitreo dell’occhio) una spiccata ipotonia-ipostenia a livello distale, la DM2 presenta al contrario una distribuzione più prossimale del deficit muscolare. L’approccio riabilitativo nei pazienti distrofici sarà di tipo multidisciplinare con il coinvolgimento di un’equipe di professionisti quali neuropsichiatra infantile, cardiologo, pneumologo, chirurgo ortopedico e fisioterapista. In tale contesto il ruolo fisioterapico vedrà la proposta di un progetto riabilitavo costituito sia da esercizi passivi che da esercizi attivi. Gli esercizi passivi saranno caratterizzati da manovre di stretching dell’ileo-psoas, del retto-femorale, del tensore della fascia lata, del tricipite femorale e del gruppo di flessori del ginocchio (ischiocrurali e gracile) negli arti inferiori, mentre negli arti superiori verranno coinvolti i gruppi muscolari agenti a livello di avambraccio, dita e polso: la ripetizione (5-10 volte) ed il mantenimento della posizione di massimo allungamento muscolare per un periodo di 15-30 secondi sono fattori implicati nella prevenzione e nel contenimento delle retrazioni muscolari. Nella proposta di esercizi attivi devono esser privilegiate attività fisiche ad intensità moderata al fine d’impedire un eccessivo affaticamento muscolare. Infine l’utilizzo di ortesi può contribuire nella prevenzione delle retrazioni muscolari e nella produzione di performance motorie qualitativamente migliori. Il secondo gruppo di patologie neuromuscolari in ordine di incidenza nella popolazione pediatrica è costituito dalle SMA (1/10.000 nati vivi), conosciute anche come Atrofie Muscolari Spinali. Il processo fisiopatologico alla base di queste malattie è rappresentato dalla progressiva perdita dei motoneuroni delle corna anteriori del midollo spinale a causa di una mutazione autosomica recessiva del gene SMN1 (“Survival Motor Neuron”), localizzato nel cromosoma 5. La degenerazione comporta una spiccata ipotonia-ipostenia muscolare, a cui si associa una riduzione della motricità spontanea. Esistono 3 forma di SMA, le quali vengono classificate in ordine decrescente di disabilità e precocità nell’outcome. La SMA di tipo I, chiamata anche Sindrome di Werding-Hoffman, è la forma a prognosi peggiore: la patologia si manifesta tra la nascita ed il sesto mese con l’impossibilità di acquisizione di qualsiasi competenza antigravitaria e problematiche sul piano respiratorio ed alimentare. Le SMA di tipo II sono caratterizzate da un outcome più tardivo che consentirà a questi paziente di apprendere basilari competenze posturali, come ad esempio la posizione seduta ed in rari casi la statica eretta mediante l’utilizzo di ausili: in questi bambini permangono tuttavia le problematiche respiratorie, le quali rappresentano globalmente la principale causa di morte. Le SMA di tipo III sono considerate forme “più lievi” ad outcome tardivo (generalmente tra l’infanzia e l’adolescenza): il processo degenerativo di queste forme non impedisce, nella maggior parte dei casi, l’acquisizione della funzione cammino. Oltre alle patologie descritte è opportuno precisare che il gruppo delle malattie neuromuscolari è estremamente vasto e che all’interno di esso risiedono anche sindromi più rare non necessariamente di natura genetica (es. Miopatie acquisite).
Con il termine di paraparesi spastica ereditaria (o sindrome di Strumpell-Lorrain) intendiamo raggruppare una serie di patologie neuromotorie rare (incidenza 1/20.000 circa di media) ad eziologia genetica variabile (X-linked, autosomico recessivo o dominante) che sono principalmente caratterizzate da rigidità, spasticità ed ipostenia negli arti inferiori: si suddividono dal punto di vista nosologico in “forme pure”, caratterizzate prevalentemente dal deficit motorio ed in “forme complesse” nella quale sono presenti disturbi associati. Pur assomigliando clinicamente alle diplegie spastiche, il processo fisiopatologico conferisce a tali malattie un quadro degenerativo piuttosto che esitale: con il trascorrere degli anni il cammino sarà qualitativamente peggiore, anche se il pattern varia notevolmente a seconda della tipo di mutazione genetica. Oltre alle problematiche motorie si possono riscontrare altri sintomi come disabilità intellettiva, epilessia, neuropatia motoria e sensitiva, alterazione della sensibilità propriocettiva e danni al nervo ottico. Il quadro clinico è determinato a livello biologico da un’alterazione del trasporto proteico assonale che risulta massimale nei neuroni più lunghi, localizzati ad esempio a livello di fascicolo gracile e vie cortico-spinali: tale fenomeno è strettamente correlato alla mutazione genetica a valle. Nella clinica risulta fondamentale la diagnosi differenziale con altre patologie grazie all’RM ed all’indagine genetica. Nella riabilitazione la fisioterapia assume un ruolo fondamentale sia con l’utilizzo di tecniche di stretching che con l’esercizio attivo per contrastare la riduzione di forza. Il trattamento farmacologico si basa essenzialmente sulla cura dei sintomi e non è al momento disponibile una reale cura risolutiva.
€ 40,00
€ 32,81
€ 35,37
€ 33,90




























