







Il ruolo delle funzioni cognitive: sviluppo normotipico e RTT a confronto
La sindrome di Rett (RTT) è una malattia neurologica che colpisce quasi esclusivamente soggetti di sesso femminile. Fu identificata nel 1966 da un medico austriaco, Andreas Rett, recentemente scomparso, ma soltanto nel 1983 venne resa nota ai clinici e a ricercatori di tutto il mondo attraverso una pubblicazione in inglese scritta da alcuni neurologi europei, i più noti dei quali erano lo svedese Hagberg e il francese Aicardi. Ci vollero, quindi, ben diciassette anni prima che la scoperta di Andreas Rett fosse divulgata nel panorama scientifico internazionale [1].
Come giovane pediatra, Andreas Rett scelse di specializzarsi in bambini con disabilità intellettive. A partire dalla metà degli anni 1950, iniziò a seguire da vicino una popolazione clinica di oltre 6.000 bambini che soffrivano di vari tipi di danni cerebrali. Fu allora che si accorse che 22 delle bambine in osservazione presentavano gli stessi sintomi insoliti con la stessa storia clinica: il loro parto era avvenuto nella norma; avevano raggiunto traguardi di sviluppo precocemente, in alcuni casi anche troppo precocemente. Nove bambine avevano avuto qualche difficoltà nella deglutizione di liquidi nei primi mesi ma la maggior parte apparivano in buona salute fino ad almeno nove mesi di età momento in cui entravano in un periodo di declino funzionale che colpiva il corpo, la mente e lo spirito. La loro capacità di stare in stazione eretta era compromessa. Le bambine che riuscivano a deambulare soffrivano di distonia e non erano quindi in grado di controllare il ritmo della loro andatura. Tutte le ragazze avevano un volto inespressivo con uno sguardo vuoto e poco interesse a interagire con il loro ambiente, compresi i loro genitori. La minoranza, che aveva imparato qualche parola mono- e bi-sillabica, aveva perso anche questo vocabolario rudimentale entro l’inizio del terzo anno di vita. Il segno più saliente, tuttavia, riguardava i movimenti stereotipati che interessavano le mani delle ragazze: da movimenti quali graffiare, accarezzare a battere e sbattere le mani sempre lungo la linea mediana in maniera incostante e limitata.
Rett allora definì questa nuova sindrome attraverso nove caratteristiche obbligatorie: 1) prevalenza del sesso femminile, 2) con manifestazioni di appiattimento affettivo (atimia), 3) afasia, 4) stereotipie delle mani, 5) iperreflessia, 6) aumento della spasticità nel tono, 7) andatura atassica, 8) tendenza a sviluppare convulsioni, 9) deficit cognitivo profondo.
Rett pubblicò il suo rapporto in tedesco nel 1966. Alcuni documenti isolati poi apparirono nel corso del decennio successivo, in cui venivano descritte ragazze con le caratteristiche che si sovrapponevano a quelle già da lui esposte. Ma quando il neurologo pediatrico svedese Bengt Hagberg lo incontrò ad un congresso medico, si rese conto che il disordine che lui e i suoi coautori avevano definito “una progressiva sindrome di autismo, demenza, atassia, e la perdita di utilizzo funzionale delle mani nelle ragazze,” era lo stesso già descritto da Rett.
Hagberg allora battezzò il disturbo con il nome di “Sindrome di Rett” nel 1983, in un documento che descriveva 35 pazienti provenienti da Francia, Portogallo e Svezia [2].
Da quando venne successivamente pubblicato nella rivista “Annals of Neurology”, la consapevolezza della sindrome aumentò rapidamente.
Nel 1992, nel laboratorio di Adrian Bird, il gene MeCP2 fu originariamente scoperto come un fattore in grado di legare le citosine metilate seguite da guanina (CpG) e successivamente in grado di legare tutte le citosine metilate. Pur non avendo ancora un quadro chiaro dell'esatta cascata molecolare con cui la disfunzione di MeCP2 conducesse a fenotipi neuropsichiatrici, era noto che la perdita o la presenza di MeCP2 potesse determinare migliaia di cambiamenti nell'espressione genica [3].
Nel 1999, il laboratorio di Zoghbi identificò mutazioni nel gene MECP2 in pazienti con RTT e la mappatura di esclusione identificò la regione candidata Xq28, includendo da quel momento il sequenziamento del gene MECP2 nella diagnosi [4].
Con la scoperta del gene vennero ampliati gli studi oltre la RTT classica anche a forme atipiche, quando si osservò che alcuni individui presentavano molte delle caratteristiche cliniche della RTT, come la regressione, pur non adattandosi completamente ai comportamenti osservati nella forma classica. Vennero così definite le varianti della RTT [5]. Nel 2004 John Christodolou individuò la mutazione del gene CDKL5 sul cromosoma X che corrispondeva alla variante ad esordio precoce [6] o sindrome di Hanefeld, dal nome di Markolf Hanefeld, che nel 1984 per primo ne descrisse il quadro clinico. Nel 2005 è stato identificato sul cromosoma 1 il gene NTNG1 coinvolto nella patogenesi di una RTT-like [7] [8]. Nel 2008 venne invece scoperto il gene FOXG1 responsabile della variante congenita [9]. L’ultimo gene recentemente scoperto implicato anch’esso nell’eziologia di una RTT-like, è il gene MEF2C, localizzato sul cromosoma 5 [10] [11] [12].
Tra le forme atipiche possiamo trovare la “Zappella Variante” caratterizzata da anomalie cliniche più lievi e dall'apparenza di una certa capacità di linguaggio; la "variante di Hanefeld", correlata al gene CDKL5, in cui vi è un inizio patognomonico di convulsioni; la "variante congenita", correlata al gene FOXG1, con insorgenza dei sintomi dopo la nascita.
Tutte queste forme hanno diversi gradi di gravità in termini di comorbidità, comportamento, prognosi e coinvolgimento del sistema nervoso autonomo [5].
La prima descrizione di RTT nella letteratura medica inglese è stata riportata da Hagberg e colleghi nel 1983, quando è stata stabilita la prima serie di criteri clinici [4].
Per tenere conto però anche dei casi atipici emergenti, era necessaria una semplificazione della diagnosi e inizialmente, nel 1994 furono sviluppati criteri di supporto per comprendere l'intera gamma di manifestazioni cliniche [13]. Nonostante ciò, era necessario un requisito per rivedere i criteri e nel 2002 venne pubblicata una nuova versione volta ad unire tutte e tre le diagnostiche esistenti riconoscendo RTT classica e RTT atipica come due entità separate. Infine, nel 2010, venne definita un’ulteriore serie di criteri per definire e semplificare ulteriormente la diagnosi: i criteri di Neul sono ora i criteri diagnostici più comunemente utilizzati per l'identificazione di nuovi casi di RTT [4].
Nel 1994 la RTT veniva posta come diagnosi differenziale del disturbo autistico secondo quanto prevedeva il Manuale Diagnostico e Statistico de Disturbi Mentali IV (DSM IV) inquadrandolo nei “Disturbi Pervasivi dello Sviluppo” insieme appunto al disturbo autistico, il disturbo disintegrativo della fanciullezza (o di Heller), di Asperger e al disturbo pervasivo dello sviluppo non altrimenti specificato [14]. Attualmente con il DSM-V, questi sottotipi sono stati riuniti in un’unica categoria denominata “Disturbi dello Spettro Autistico”, ad eccezione della RTT che è stata invece posta tra i disturbi neurologici. L’unificazione dei diversi disturbi pervasivi dello sviluppo in un’unica categoria è la conseguenza di studi scientifici che hanno dimostrato come la distinzione in sottotipi diagnostici non sia coerente nel tempo e come le differenze nelle abilità sociali e cognitive dei sottogruppi si caratterizzino meglio in termini di un continuum [15].
Negli ultimi dieci anni, geni di malattie, fenotipi clinici supplementari e ulteriori ridefinizioni diagnostiche sono stati associati con la sindrome, nuove terapie sono in fase di sperimentazione in studi clinici ed è stata stabilita una maggiore comprensione generale dell’intero spettro clinico della malattia per consentire una migliore opzione di trattamento e la qualità della vita per i pazienti con RTT [4].
La malattia è diffusa in tutto il mondo e, da alcune ricerche epidemiologiche, si pensa che colpisca la popolazione femminile con rari casi segnalati nei maschi. Presenta una frequenza che va da 1:10000 a 1:18000, a seconda delle zone territoriali studiate: ciò significa che nelle bambine è molto più comune della fenilchetonuria, una nota malattia congenita del metabolismo che può essere diagnosticata esaminando il sangue del neonato.
Nell’autunno del 1986 vi erano circa 1150 casi noti di RTT in tutto il mondo. A quel tempo in Svezia le ragazze affette erano circa 80, di età compresa fra i 2 e i 40 anni [1].
Il primo studio epidemiologico è stato effettuato da Hagberg nel 1985, che ha trovato una
prevalenza nel sud-ovest della Svezia di 0,65 per 10.000 donne di età compresa tra 6-17 anni, che è circa il doppio di quello della fenilchetonuria nella stessa area [16].
Gli studi effettuati in altre regioni europee e paesi prima l’identificazione di MECP2 ha riferito che la prevalenza è stata di 1 per 10.000 donne in Svezia, e 0,80 per 10.000 femmine
sotto i 15 anni in Scozia [17]. La prevalenza in Australia è stata simile a quella in Svezia e Scozia (0,72 per 10.000 femmine) [18]. Tuttavia, i tassi di prevalenza riportati per le diverse regioni in Giappone variano notevolmente: 0,67 per 10.000 femmine di età compresa tra 6-17 anni (0,50 per 10.000 femmine se limitata alla RTT di tipo classico) nel quartiere Tama di Tokyo; 0,50 per 10.000 femmine di età compresa tra 6-14 anni (0,40 per 10.000 femmine se limitato alla RTT non incluse le varianti) a Tokyo metropolitana; 0,36 per 10.000 femmine sotto i 16 anni (0,37 per 10.000 femmine di età compresa tra 7-15 anni) nella prefettura di Tokushima, e 0,22 per 10.000 donne di età compresa tra 6-14 anni nella prefettura di Fukui [19]. In uno studio più recente, tra le donne di età compresa tra 0 e 18 anni nel Nord Dakota, è stata trovata una prevalenza della RTT di 0,505 per 10.000 [20]. In tutti questi studi, i criteri di diagnosi dipendono dalla consapevolezza relativa alla malattia e il follow-up di crescita iniziale e di sviluppo. La diagnosi è ostacolata dalla carenza di informazioni riguardanti la patogenesi e la mancanza di marcatori biologici specifici Rett sindrome. Inoltre, a causa di una descrizione della RTT disponibile nella letteratura medica in lingua inglese solo dal 1983, è altamente probabile che molti professionisti della salute non erano pienamente consapevoli della RTT, quando i primi studi epidemiologici furono attuati [21].
Attualmente, attraverso studi epidemiologici efficaci, è stato attestato che la prevalenza di questo disordine dello sviluppo neurologico, che colpisce principalmente ragazze, è di 1/8500 [22]. In particolare, nelle ragazze di età inferiore ai 12 anni con RTT, l’incidenza è stimata a 1/9000 [23].
Recentemente è stato identificato che, per la maggior parte delle mutazioni, indipendentemente dalla gravità iniziale della manifestazione clinica, la gravità peggiora progressivamente con l'età [4]. Con il passare del tempo infatti, oltre alla perdita progressiva delle abilità in precedenza acquisite, come l'uso intenzionali delle mani, il linguaggio, la deambulazione e le capacità comunicative cominciano anche a manifestarsi convulsioni, problemi cardiaci e respiratori. Al giorno d'oggi, la terapia farmacologica è essenzialmente sintomatica e mirata a trattare sintomi come convulsioni, disturbi del sonno e del comportamento [24].L’aspettativa media di vita di una persona con RTT si stima possa superare i 47 anni. La prevenzione ed il controllo, affiancati da una corretta alimentazione e da terapie farmacologiche adeguate, sono in grado di garantire condizioni di buona salute [25].
Dal momento che la ricerca di geni nei primi anni ’80 e ’90 era ancora scoraggiante, si sperava di trovare i pazienti con anomalie cromosomiche, che potessero restringere la regione candidata sul cromosoma X. I primi studi di diversi laboratori furono focalizzati su un possibile sito fragile Xp22 in Rett, ma si rivelarono essere comuni nella popolazione generale. Quella fu la prima falsa pista nella genetica Rett.
Successivamente vennero individuate due famiglie, ognuna delle quali aveva un paio di sorellastre con la madre come genitore in comune affette, che sostenevano il concetto di ereditarietà X-linked dominante. Si ipotizzò che la madre fosse portatrice di un allele mutante, ma che fosse protetta a causa della favorevole inattivazione del cromosoma X, che si rivelò poi essere il caso per una delle madri [26].
Le famiglie aiutarono a restringere la regione candidata al terzo braccio del cromosoma X in base alle coppie di sorellastre che condividevano questa regione del loro cromosoma X.
Si pensò di aver avuto un colpo di fortuna quando si identificò un paziente con Rett e una X: Traslocazione 3, ma questo si rivelò essere la seconda falsa pista: la clonazione del punto di rottura svelò che nessun gene era stato interrotto dalla traslocazione. La speranza rinacque quando venne identificata una famiglia con due cugine secondogenite con la RTT, che erano imparentate per via materna e condividevano una bisnonna. Escludendo la mappatura genetica, tuttavia, venne dimostrato che queste due cugine affette non condividevano alcuna parte del cromosoma X della bisnonna. Rivelazione che fu una grande delusione che quasi escluse l’ipotesi X-linked.
In seguito, venne identificata una famiglia che aiutò a restringere il campo sulla regione Xq con i dati di esclusione di una mappatura da una quarta famiglia ulteriormente ristretti alla regione in Xq28 [27].
Nel 1999, dopo 16 anni di mappatura e sequenziamento sistematico dei geni candidati, fu finalmente scoperta la causa della RTT: mutazioni responsabili di perdita di funzione nel gene che codifica per il metil-CpG binding protein 2 (MECP2) [28].
Tutte le ragazze che aiutavano a restringere il campo di studio sulla regione candidata sul cromosoma X, avevano avuto una perdita di funzione delle mutazioni del MECP2. La ragazza con l’inversione aveva avuto una mutazione MECP2 sul cromosoma X non-invertito in cui l’inversione era stata quindi una coincidenza. Le due mezze cugine che non condividevano alcun DNA avevano effettivamente mutazioni in MECP2, ma il disturbo era sporadico in entrambe le ragazze (le probabilità che ciò accada due volte nella stessa famiglia sono veramente basse). Questo ci fa capire come sequenze di anomalie nei pazienti devono essere attentamente interpretati nel contesto di tutte le possibilità e il resto del genoma.
Oggi sappiamo che il 95% degli individui con RTT classica presenta mutazioni in MECP2. Il restante 5% può avere mutazioni nelle regioni di controllo regolatorie [3].
L'inattivazione del cromosoma X (XCI) esercita una notevole influenza sulla manifestazione fenotipica di una mutazione legata al cromosoma X nelle femmine eterozigoti modificando stocasticamente la proporzione di due tipi di cellule, una con il cromosoma X attivo di derivazione paterna (XP) e l'altro con il cromosoma X attivo di origine materna (XM). Apparentemente, la gravità dei sintomi sembra dipendere dalla percentuale di cellule che hanno inattivato il normale cromosoma X.
XCI, un evento di sviluppo che si verifica nella differenziazione delle cellule embrionali, è un meccanismo di compensazione del dosaggio tra femmine XX e maschi XY, in mammiferi che equalizzano i prodotti genici legati all'X, attraverso il silenziamento di uno dei cromosomi X nelle cellule femminili. Nella maggior parte dei pazienti con RTT, il modello di inattivazione X è stato determinato nei linfociti del sangue periferico. Sebbene apparentemente la malattia sia causata da una mutazione con perdita di funzione di MeCP2, è comunque ancorata l’idea dell’eredità X-linked dominante con la letalità maschile. Questa insolita espressione fenotipica può essere spiegata dal fatto che il gene MECP2 legato all'X è soggetto all'XCI. È stato infatti ha dimostrato che la natura stocastica dell'XCI amplia la variabilità fenotipica nella RTT classica [29]. Inoltre, l'elevata incidenza di XCI casuali nei pazienti con RTT classica ha portato a concludere che è l'espressione del mutante MECP2, in un’elevata percentuale di cellule dovuta a XCI, a condurre le femmine eterozigoti alla manifestazione della malattia [30].
Sebbene ci sia ancora molto da chiarire, sembra probabile che sia la mutazione MECP2 sia l'XCI casuale siano responsabili della patogenesi della RTT [29].
I meccanismi fisiopatologici alla base di RTT sono ancora da chiarire. Si pensa che mutazioni inattivanti del gene MECP2 abbiano a che fare con la perdita parziale o completa della capacità di silenziare i promotori di geni che non sono più presenti o non sono necessari in particolari tipi di cellule [31], con conseguente anormalità nei sistemi di numerosi neurotrasmettitori (compresi colinergici, dopaminergici, glutaminergici, serotoninergici e trasmissione GABAergica) e fattori trofici (incluso il fattore di crescita nervosa e il fattore neurotrofico di derivazione cerebrale). Studi citometrici immuno-reattivi hanno dimostrato che la presenza di MeCP2 nei neuroni differenziati risulta ridotta nella RTT [32].
L'esatto meccanismo con cui la perdita MECP2 abbia a che fare con le caratteristiche cliniche distintive di RTT rimane incerta; si suppone che anormali risposte corticali sinaptiche glutamatergiche e eccitatorie risultano connesse ad un eccesso di inibizione e deficit di plasticità neuronale. Mutazioni MECP2 si verificano di solito de novo: in più del 99% dei casi queste mutazioni si verificano sporadicamente, in meno dell’1% sono ereditate da un genitore; casi familiari provengono da mutazioni ereditate da madri sane o lievemente affette o da mosaicismo gonadico. La maggior parte dei pazienti è quindi eterozigote per la mutazione MECP2, portando una copia normale e una copia mutata del gene.
La maggior parte delle mutazioni de novo si verificano quasi esclusivamente nel gamete paterno [33].
Anche se la maggior parte dei pazienti RTT presentano mutazioni del gene MECP2, mutazioni di altri geni sono associati a RTT, tra cui chinasi-ciclo dipendente 5 (CDKL5), Forkhead box protein G1 (FOXG1), fattore di attivazione miocita-specifico 2C (MEF2C), e fattore di trascrizione 4 (TCF4) [34] [35].
MECP2. MECP2 è una proteina pleiotropica abbondantemente espressa nel cervello. È un membro della famiglia del dominio di legame metilico (MBD) ed è un modulatore epigenetico chiave nel cervello, che controlla l’architettura della cromatina e l’espressione genica attraverso il legame al DNA metilato. Le mutazioni del MECP2 sono state identificate come la causa genetica primaria di RTT [28].
MECP2 svolge un ruolo importante nello sviluppo e nella funzione neuronale, attraverso la sua capacità di legarsi al DNA metilato e interagisce con la co-regolazione dei partner proteici di trascrizione nelle cellule neuronali. Il meccanismo generale di sviluppo neuronale attraverso la mediazione di MECP2 non è ancora chiaro, tuttavia sappiamo che le mutazioni in MECP2 possono mettere in pericolo lo sviluppo neuronale. Ci sono ampi studi in vivo che sottolineano l’importanza di questo gene nello sviluppo neuronale, ramificazioni dendritiche, e la morfologia del cervello che confermano la correlazione genotipo – fenotipo tra MECP2 e RTT. Stime recenti riconoscono che circa il 95% dei casi di RTT classica e il 75% dei casi di RTT atipica hanno mutazioni in MECP2 [36].
Ad oggi oltre 900 mutazioni uniche sono state identificate in MECP2 con 518 effetti patogeni o potenzialmente patogeni, 206 benigni o potenzialmente considerabili come patogeni e 211 varianti di significato sconosciuto (VSS) (Grafico 1).

Grafico 1. Distribuzione della patogenecità delle varianti in MECP2, CDKL5 E FOXG1. In grigio sono indicate le VSS; in blu le mutazioni benigne o potenzialmente benigne; in arancione le mutazioni patogene o potenzialmente patogente.
In particolare, sono state identificate mutazioni puntiformi, inserzioni, duplicazioni, delezioni piccole o grandi in quasi tutte le parti del gene. La maggior parte delle mutazioni è stata trovata in otto poli-nucleotidi singoli poli-morfismi hotspot come mutazioni missenso e senza senso: T158M; R255X; R168X; R306C; R294X; R270X; R133C [37]. Queste mutazioni rappresentano circa il 70% di tutte le mutazioni e R168X è la più comune; le delezioni C-terminale rappresentano l'8% e le grandi delezioni un altro 5% [36].
Nonostante l'ampio spettro di mutazioni MECP2 e la varietà della gravità del fenotipo, non ci sono chiare correlazioni genotipo-fenotipo [38] [36].
Alcuni studi in letteratura hanno proposto una correlazione tra genotipo e caratteristiche cliniche in pazienti RTT: mutazioni troncanti precoci quali R168X, R255X e R270X e grandi inserzioni e delezioni causano i fenotipi più gravi mentre mutazioni missenso come R133C e R306C e mutazioni tardive come quelle di R294X e altre all'estremità 3', sono associate con il fenotipo più lieve. Nonostante questo, le variazioni fenotipiche si verificano comunemente tra pazienti con la stessa mutazione [39].
Sono state proposte alcune possibili spiegazioni come differenze di inattivazione del cromosoma X e la presenza di una seconda mutazione genica che agisce modificando o migliorando l’esito del fenotipo [33].
La gravità del fenotipo RTT dipende dalla natura della mutazione e quasi ogni delezione esonica nella proteina è considerata patogena. L’analisi strutturale di MeCP2 ha evidenziato tre domini principali all'interno della proteina: il methyl binding domain (MBD), il dominio di repressione trascrizionale (TRD) e il dominio del segnale di localizzazione nucleare (NLS). La maggior parte delle mutazioni che colpiscono questi domini sono patogeni rispetto a mutazioni che rientrano in altre zone della proteina come le regioni C-terminale e N-terminale. Fino al 60% di tutte le mutazioni patogene si trovano all'interno di MBD e di TRD con 8 principali mutazioni disperse lungo queste regioni che rappresentano circa il 47% di tutte le mutazioni.Troncature e mutazioni missenso costituiscono la maggioranza delle mutazioni delle quali 104 con effetti patogeni di tutte le 110 troncature e 70 di tutte le 252 mutazioni missenso. Le mutazioni introniche sono meno comuni con 14 mutazioni patogene su un totale di 43. Varianti nella 3 ' UTR e 5 ' UTR comprendono una parte considerevole delle varianti totali (85, 60, e 5, rispettivamente); Tuttavia, nessuno di queste ha una prova funzionale di patogenicità [40] (Grafico 2).

Grafico 2. Numero di varianti MECP2.
Mentre la maggior parte dei pazienti RTT presenta mutazioni / delezioni localizzate nel gene MECP2, circa il 5% dei pazienti con RTT classica e il 25% dei pazienti con RTT atipica presenta una mutazione MECP2 negativa [41].
CDKL5. I primi referti in cui fu riportato che le mutazioni della ciclina chinasi-dipendente like5 (CDKL5) erano state associate con RTT, furono pubblicati nel 2004 e da allora furono riportati molti altri casi fino a raggiungere un totale di 499 individui (368 femmine e 66 maschi). Di questa coorte, 209 individui avevano mutazioni patogene,177 femmine e 31 maschi, con 159 individui diagnosticati con RTT, 155 femmine e 4 maschi [40].
Ad oggi, oltre 255 mutazioni uniche sono state identificate con CDKL5, di cui 164 mutazioni con effetti patogeni o potenzialmente patogeni, 65 con effetti benigni o probabilmente benigni.
Le caratteristiche cliniche più comuni comprendono:
Inoltre, vari aspetti del viso sono stati descritti in pazienti con CDKL5 tra cui una fronte ampia, occhi infossati, naso ben pronunciato, dita affusolate, e labbra carnose.
Una valutazione fenotipica completa di un’ampia coorte di pazienti con CDKL5 è stata recentemente pubblicata con l’obiettivo di determinare se questi pazienti in realtà rientrano nella gamma di RTT. Lo studio di Fehr et al. ha dimostrato che quasi il 25% della loro coorte di 86 pazienti con mutazioni del CDKL5 non ha soddisfatto i criteri Neul per le convulsioni ad insorgenza precoce della sindrome di RTT. Gli autori hanno quindi proposto che i pazienti con mutazioni nel CDKL5 dovrebbero essere considerati un’entità separata dalla RTT, piuttosto che essere una variante del disturbo, e suggeriscono che dovrebbe essere noto come il disturbo CDKL5 [42].
FOXG1. Un piccolo numero di pazienti con diagnosi di RTT presentano mutazioni sul gene della proteina Forkhead box G1 (FOXG1) e con diagnosi della variante congenita di RTT. Nel 2008, sono state identificate due mutazioni del gene FOXG1 in due individui a cui era stata diagnosticata la forma congenita di RTT. Ad oggi sono stati riportati 59 pazienti con mutazioni patogene di FOXG1, 36 femmine e 23 maschi dei quali solo 27 con diagnosi di RTT atipica (20 femmine e 7 maschi) [40]. 44 mutazioni uniche sono state identificate con FOXG1 di cui 41 ad effetti patogeni o potenzialmente patogeni, 2 ad effetti benigni o probabilmente benigni e 1 VOUS (varianti di significato sconosciuto).
FOXG1 codifica un repressore trascrizionale cerebrale specifico che è essenziale per lo sviluppo cerebrale precoce. Tuttavia, come è avvenuto per le mutazioni di CDKL5, ci sono caratteristiche che si vedono comunemente nei pazienti con mutazioni di FOXG1, come agenesia o ipoplasia del corpo calloso, che non sono state riscontrate in individui con mutazioni di MECP2 o CDKL5, e di conseguenza, è stato suggerito che le mutazioni in FOXG1 possano causare un’entità clinica distinta da RTT [4].
Dal primo rapporto del 1966, la RTT è stata diagnosticata solo su base clinica. Tuttavia, nel 1999, il laboratorio Zoghbi identificò mutazioni nel gene MECP2 in pazienti con Rett e il sequenziamento del gene MECP2 venne incluso nella diagnosi [28].
Raramente mutazioni in MECP2 sono state identificate in disturbi isolati dello spettro autistico e ritardo mentale sindromico. Inoltre, non tutti i pazienti con una diagnosi clinica di RTT hanno una mutazione in MECP2 (o qualsiasi altro gene noto), e sono quindi classificati come mutazione negativa. Infatti, circa il 5% dei pazienti che soddisfano i criteri clinici diagnostici per RTT classica non hanno mutazioni in MECP2, con il numero di pazienti gene-negativi che risulta più alto nei casi di RTT atipici. Inoltre, i pazienti con un fenotipo clinico che si sovrappone con RTT, ma non soddisfano i criteri diagnostici clinici rivisti, sono chiamati Rett-like, e includono la sindrome di Angelman, Pitt - sindrome di Hopkins, e alcune encefalopatie epilettiche. Date queste informazioni, è ormai accettato che la diagnosi di RTT si basa sia su scoperte cliniche che molecolari [4].
Non tutti i sintomi sono presenti inizialmente, ma piuttosto appaiono in fasi (Figura 1). I pazienti con RTT sembrano svilupparsi normalmente fino a 6-18 mesi di età. Il bambino con RTT raggiunge apparentemente le pietre miliari del caso, tra cui la capacità di camminare, e alcuni pazienti iniziano a dire qualche parola. Un indicatore precoce di coinvolgimento neurologico è la decelerazione della crescita della circonferenza cranica, con conseguente microcefalia dal secondo anno di vita. Con l'inizio della stagnazione dello sviluppo, la microcefalia acquisita è accompagnata da ritardo generale della crescita, perdita di peso, e una postura debole, causata da ipotonia muscolare.
Come la sindrome progredisce, i pazienti perdono l'uso funzionale delle loro mani e si sviluppano movimenti stereotipati come hand wringing or washing movements.
La perdita della socialità e del linguaggio si manifestano insieme a irritabilità e comportamenti autolesionisti. Altre caratteristiche autistiche che compaiono sono il volto inespressivo, ipersensibilità ai suoni, mancanza di contato oculare, indifferenza all’ambiente circostante e mancanza di regole sociali. Il deterioramento mentale è accompagnato da perdita di coordinazione motoria e lo sviluppo di un’andatura atassica. Il primo disturbo del sistema autonomo riguarda l’iperventilazione durante la veglia. La maggior parte delle ragazze con RTT soffrono di anomalie respiratorie aggiuntive, tra cui trattenere il respiro, aerofagia, l'espulsione forzata di aria e saliva, apnea. Una delle caratteristiche più comuni è il verificarsi di crisi epilettiche, di cui i tipi più comuni sono quelle tonico-cloniche parziali [43]. Le crisi epilettiche tendono a diminuire con l’età, dopo l'adolescenza e in età adulta, e presentano minori problemi dopo i quarant'anni. Un miglioramento della componente sociale si verifica a volte tra 5 a 10 anni di età. Pur avendo un appetito normale, i pazienti continuano a perdere peso e molti soffrono di osteopenia, scoliosi, e rigidità andando avanti on l’età. Anomalie del comportamento durante questa fase di post regressione includono digrignamento dei denti, urla, umore basso, ed episodi di ansia indotte da eventi esterni angoscianti [44]. I pazienti presentano grave deterioramento motorio, rigidità generalizzata, distonia, peggioramento della scoliosi. La maggior parte delle ragazze con RTT perde la mobilità, ed è spesso sulla sedia a rotelle durante l'adolescenza. Ulteriori anomalie del sistema autonomo includono ipotrofia, piedi freddi e blu, stipsi grave, disfunzioni orofaringee, e anomalie cardiache, tra cui tachicardia, e bradicardia sinusale. Quando i pazienti invecchiano spesso sviluppano caratteristiche parkinsoniane [45].
La condizione raggiunge un plateau e alcuni pazienti sopravvivono fino alla sesta o settima decade di vita in una condizione fisica gravemente debilitati.
![Figura 1. Dopo un periodo di sviluppo normale, una bambina dall'aspetto sano cade in una fase di stagnazione dello sviluppo, seguita da un rapido deterioramento, perdita del linguaggio acquisito, e la sostituzione di un utilizzo funzionale delle mani con stereotipie incessanti. I pazienti sviluppano anche anomalie nel comportamento sociale. La condizione peggiora con la perdita delle abilità motorie e un deterioramento cognitivo profondo. Inoltre, i pazienti soffrono di ansia, attacchi epilettici e una serie di anomali autonomiche [46].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/figura-1-periodo-sviluppo-normale-fase-di-stagnazione-dello-sviluppo.png)
Figura 1. Dopo un periodo di sviluppo normale, una bambina dall'aspetto sano cade in una fase di stagnazione dello sviluppo, seguita da un rapido deterioramento, perdita del linguaggio acquisito, e la sostituzione di un utilizzo funzionale delle mani con stereotipie incessanti. I pazienti sviluppano anche anomalie nel comportamento sociale. La condizione peggiora con la perdita delle abilità motorie e un deterioramento cognitivo profondo. Inoltre, i pazienti soffrono di ansia, attacchi epilettici e una serie di anomali autonomiche [46].
Secondo i criteri diagnostici riveduti per RTT di Neul et al, nella RTT classica, sono necessari un periodo di regressione seguita dal recupero o la stabilizzazione e la presenza di quattro criteri principali e due criteri di esclusione per la diagnosi (Tabella 1).
![Tabella 1. Criteri clinici della RTT classica [36].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/tabella-1-criteri-clinici-della-rtt-classica.png)
Tabella 1. Criteri clinici della RTT classica [36].
Per quanto riguarda la decelerazione postnatale della crescita della testa, è un segno precoce che inizia tra i 2 ei 4 mesi di età. Tuttavia, non si trova in tutti i pazienti con RTT tipica, per questo motivo è stata eliminata dai criteri necessari, ma è considerata oggi un loro preambolo, come una caratteristica che dovrebbe sollevare il sospetto per la diagnosi.
Il decorso della malattia, nella sua forma classica, è caratterizzato da quattro fasi con un apparentemente normale e precoce sviluppo psicomotorio nei primi 6 mesi di vita (Tabella 2).
![Tabella 2. Stadi clinici nella RTT classica [47].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/tabella-2-stadi-clinici-nella-rtt-classica.png)
Tabella 2. Stadi clinici nella RTT classica [47].
In alcuni casi, possono essere presenti sintomi vaghi, come ipotonia e ridotta interazione sociale. Einspieler et al. Ha valutato attentamente i video di 22 casi di Rett, esaminando i movimenti, la postura, e il comportamento durante i primi 6 mesi di vita. Gli autori hanno dimostrato una qualità anormale nei general movements (100%), protrusione della lingua (62%), rigidità posturale (58%), apertura e chiusura degli occhi asimmetrica (56%), movimenti anormali delle dita (52%), stereotipie delle mani (42%), esplosione di espressioni facciali anormali (42%), sorriso bizzarro (32%), tremore (28%), e movimenti corporei stereotipati (15%) [48].
Oltre alla forma classica, è stato riconosciuto che alcuni individui presentano molte delle caratteristiche cliniche di RTT, ma non hanno propriamente tutte le caratteristiche di questa condizione. In questo caso si parla di Rett “atipica” o “variante”: la variante del linguaggio conservato (Zappella variante), la variante congenita (Rolando variante), e la variante delle crisi precoci (Hanefeld variante). Per la diagnosi di RTT atipica o variante, sono necessari un periodo di regressione seguito dal recupero o dalla stabilizzazione e almeno due dei quattro criteri principali e cinque dei 11 criteri di supporto (Tabella 3).
![Tabella 3. Criteri diagnostici per la variante atipica di RTT [36].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/tabella-3-criteri-diagnostici-per-la-variante-atipica-di-rtt.png)
Tabella 3. Criteri diagnostici per la variante atipica di RTT [36].
Secondo i dati degli United States RTT National History Study, su un campione di 819 pazienti, 85,4% soddisfaceva i criteri diagnostici per la classica RTT e il 14,6% per la RTT atipica [49].
La Zappella variante è una variante clinica caratterizzata da regressione a 1-3 anni, fase di plateau prolungata, una compromissione dell’uso funzionale delle mani più lieve e una disabilità intellettiva meno grave. La caratteristica distintiva di questa variante è il recupero del linguaggio dopo la regressione a un’età media di 5 anni, insieme ad una rara epilessia. Nella maggior parte dei casi si trovano mutazioni in MECP2.
La Rolando variante o variante congenita è caratterizzata da un grave ritardo psicomotorio con incapacità di camminare, una severa microcefalia postnatale, regressione nei primi 5 mesi, assenza del tipico contatto oculare della RTT, anomalie del sistema autonomo come mani e piedi freddi, disturbi dei vasi periferici, anomalie della respirazione in fase di veglia, e anomalie nei movimenti quali stereotipie della lingua, movimenti a scatti degli arti. Mutazioni del gene MECP2 si trovano raramente in questa variante, che è strettamente legata invece a mutazioni in FOXG1.
La Hanefeld variante è caratterizzata da un esordio precoce di convulsioni (spasmi infantili e epilessia refrattaria mioclonica) prima della regressione, di solito prima dei 5 mesi di vita. Altre caratteristiche tipiche della RTT sono meno frequenti. Inoltre, in questa variante, mutazioni nel gene MECP2 si trovano raramente, in quanto si caratterizza per mutazioni nel CDKL5 [33].
La parola cognizione deriva dalla parola latina "cognoscere" che significa conoscere. Quando parliamo di cognitivo infatti ci riferiamo alla capacità che abbiamo noi esseri viventi di elaborare le informazioni provenienti dalla percezione, ovvero gli stimoli che ci arrivano dal mondo esterno attraverso i sensi, la conoscenza acquisita con la nostra esperienza soggettiva e le caratteristiche che ci permettono di integrare tutte queste informazioni per valutare e interpretare il mondo [50]. In psicologia con "cognitivo" infatti si intende un processo psichico/mentale mediante il quale "un organismo acquisisce informazioni sull'ambiente e le elabora a livello di conoscenze in funzione del proprio comportamento". Essa corrisponde dunque alla creazione e alla manipolazione delle rappresentazioni mentali [51].
Esistono diversi tipi di funzioni cognitive tra cui: 1) il linguaggio, 2) la rappresentazione dei gesti, 3) la memoria, 4) la rappresentazione del corpo, 5) l’identificazione degli oggetti, 6) l'attribuzione di un significato, 7) la conoscenza dello spazio, 8) le capacità di apprendimento.
Ad eccezione della memoria, tutte le funzioni sopracitate sono chiamate funzioni strumentali, in quanto possono essere definite come funzioni esecutive complementari che consentono la generazione e il controllo di comportamenti volontari diretti verso un obiettivo. Di estrema importanza troviamo la vigilanza e l’attenzione, indispensabili per “orchestrare” la melodia del pensiero dove le funzioni strumentali sono quelle che “suonano” sotto il controllo di un direttore: le funzioni esecutive [52].
Le funzioni esecutive sono le capacità cognitive necessarie a eseguire comportamenti complessi diretti a uno scopo e ad adattarsi a un range di cambiamenti e richieste dell’ambiente. Queste funzioni includono la capacità di pianificare e anticipare l’esito delle azioni come la flessibilità cognitiva, ma anche la capacità di indirizzare le risorse attentive alla soddisfazione delle richieste di eventi non routinari e le capacità di automonitoraggio e autoconsapevolezza che sono necessarie per eseguire comportamenti appropriati [53].
L’espressione «Funzioni Esecutive» (FE) è stata utilizzata per la prima volta circa trent’anni fa dalla neuropsicologa americana Muriel Lezak per descrivere quelle capacità cognitive che rendono un individuo in grado di eseguire un comportamento indipendente, finalizzato e adattivo [54].
Nel 1990 Baddeley definì il sistema delle FE come un complesso di meccanismi che consente di ottimizzare la prestazione in situazioni che richiedono la simultanea attivazione di processi cognitivi differenti. Tali funzioni appaiono particolarmente critiche quando devono essere organizzate ed eseguite risposte comportamentali complesse o nuovi programmi di azione [55].
Nel 1997 Owen le definì invece come processi responsabili di schemi cognitivo-comportamentali adattivi, elaborati in risposta a condizioni ambientali nuove o impegnative [56]. Nonostante un sostanziale accordo sulla definizione del costrutto, la natura monolitica o multicomponenziale del sistema delle FE è stata per lungo tempo argomento di dibattito fra gli studiosi. Tradizionalmente sono state considerate un sistema unitario, corrispondente a un «esecutivo centrale» in grado di controllare la selezione, l’attivazione e il mantenimento dei processi cognitivi e di modificare il comportamento in base a scopi specifici [57].
Il funzionamento dell’esecutivo centrale è stato descritto accuratamente in un modello proposto da Norman e Shallice: un modello gerarchico, secondo cui il funzionamento di processi automatici (livello 1) è controllato da schemi (livello 2). Tali processi possono essere attivati anche simultaneamente in tutte le situazioni in cui bisogna pianificare o prendere decisioni, intraprendere un’azione nuova, correggere gli errori, frenare risposte apprese ma inadeguate, affrontare una situazione complessa. In altri termini, secondo gli autori la maggior parte delle attività umane viene controllata da schemi di azioni innescati da determinati stimoli ed eseguiti automaticamente [58].
Shallice ha successivamente perfezionato queste teorizzazioni, sostenendo che i meccanismi di regolazione semiautomatici sono governati da un sistema superiore di controllo volontario e consapevole, denominato Sistema Attentivo Supervisore (SAS). Il SAS opera a un livello superiore (livello 3) senza controllare direttamente il comportamento, ma modulando i livelli inferiori del sistema attraverso l’attivazione o l’inibizione di determinati schemi [59].
È nell’ambito di tale modello che vengono delineate per la prima volta alcune sottocomponenti del sistema esecutivo, consentendo una revisione del costrutto delle FE. Proprio Shallice, infatti, descriverà alcuni processi cognitivi appartenenti al dominio esecutivo che possono risultare isolatamente compromessi: 1) l’organizzazione delle azioni in sequenze gerarchiche di mete; 2) lo spostamento flessibile dell’attenzione sulle informazioni rilevanti; 3) l’attivazione di strategie adeguate per il raggiungimento di un dato scopo; 4) l’inibizione di risposte attivate da stimoli esterni ma inadeguate [60].
Progressi più recenti della neuropsicologia cognitiva moderna hanno fornito ampia conferma empirica circa l’organizzazione multicomponenziale e modulare dei processi mentali [61].
Su questa scia prevale ormai la tendenza a considerare il dominio delle FE scomponibile in funzioni cognitive parzialmente differenziabili, in particolare verrebbero considerate come dei processi higher level, coinvolti nel controllo e nella regolazione di processi cognitivi lower level. Inoltre, i diversi esiti sia cognitivi che comportamentali di lesioni a carico di specifiche aree della corteccia prefrontale [62] — principale sede anatomica delle FE [63] —, hanno suggerito la distinzione tra aspetti esecutivi «caldi» e «freddi» [64]):
Attualmente, malgrado vi sia ormai accordo tra gli studiosi sulla declinazione multicomponenziale del costrutto delle FE, si conoscono più di 30 definizioni operative di FE, a seconda del maggiore o minore rilievo che viene dato ai sottoprocessi che le compongono. E sebbene non sia stato raggiunto un accordo in letteratura sul numero di fattori individuati, le FE a cui si è più spesso fatto riferimento comprendono: 1) l’attivazione e la regolazione dei processi attentivi volontari, 2) le capacità di astrazione e di ragionamento, 3) la programmazione di strategie per la risoluzione di un compito (problem solving), 4) la flessibilità cognitiva, 5) le abilità di pianificazione, 6) l’inibizione di comportamenti automatici per far fronte a eventi nuovi e inattesi, 7) la memoria di lavoro e la regolazione del comportamento emotivo.
Tutte queste componenti sono a loro volta influenzate da funzioni neuropsicologiche di base quali: 1) l’attenzione, 2) la memoria, 3) l’intelligenza verbale e 4) l’intelligenza di performance [65].
La pianificazione è un insieme di attività cognitive che anticipano e regolano il comportamento, e consentono di eseguire una sequenza di azioni al fine di raggiungere una meta. In altri termini ci si può riferire alla pianificazione come a un’attività simbolica che consiste nel prefigurare una sequenza di azioni sufficiente per raggiungere un obiettivo [66] [67]. Ai fini di un’efficace riuscita nel processo di pianificazione, infatti, è necessario anticipare e «tenere a mente» le conseguenze di un’azione sulle altre: nel risolvere un compito, un soggetto deve essere in grado di costruire una mappa mentale del percorso corretto, attraverso l’individuazione anticipata dell’unica soluzione del problema [68].
Occorre sottolineare che per pianificare è necessario, innanzitutto, dirigere l’attenzione e focalizzarla sul problema in modo funzionale in modo da mantenere la concentrazione sul compito per periodi relativamente lunghi, mantenendo un’accurata capacità di ricezione e di elaborazione degli stimoli in ingresso. Inoltre, alla base di un processo di pianificazione efficace in particolare in contesti nuovi e complessi vi è la capacità di verificare, monitorare i piani d’azione formulati e operare costantemente la loro implementazione, al fine di potere operare eventuali modifiche e aggiustamenti in action, se richiesto dalle caratteristiche del contesto [69]. Notevole rilievo assumono anche le capacità di astrazione, ragionamento e flessibilità cognitiva: per pianificare e risolvere adeguatamente un compito è necessario essere in grado di passare rapidamente da un concetto all’altro e saper attribuire significati diversi a uno stesso concetto.
Appare evidente come tutto il sistema esecutivo necessiti del concorso e della modulazione di diverse funzioni mentali come: 1) l’attenzione, 2) l’astrazione, 3) il ragionamento, 4) la memoria di lavoro, 5) la formulazione di un piano d’azione, 6) il monitoraggio delle azioni e 7) la valutazione del risultato. La pianificazione inoltre risulta coinvolta in altri processi cognitivi superiori, che in questa sede non saranno trattati in maniera specifica, quali il problem solving e i processi di decision making [70].
Un aspetto connesso con il complesso delle funzioni esecutive, sebbene altamente specifico, funzionalmente e strutturalmente autonomo, è il movimento e la sua componente più esecutiva di monitoraggio e pianificazione del movimento.
I movimenti diretti a uno scopo dipendono dalla conoscenza del luogo in cui è collocato il corpo nello spazio, dalla direzione che si intende prendere e dalla selezione di un piano per arrivare nel luogo prescelto. Una volta che il piano è stato selezionato, deve essere mantenuto in memoria fino al momento giusto. È infine necessario eseguire correttamente i singoli atti motori per rendere il piano effettivo [69].
Questi diversi aspetti del piano motorio coinvolgono l’attività di diverse zone della corteccia cerebrale. Esistono infatti quattro aree fondamentali deputate al controllo volontario: 1) la corteccia motoria, 2) la corteccia premotoria, 3) l’area supplementare del lobo frontale e 4) la corteccia parietale posteriore. Nell’attività di queste aree si realizza una sintesi funzionale tra il controllo delle singole unità motorie e l’influenza globale della corteccia sull’attività motoria.
La corteccia motoria è un’area circoscritta del lobo frontale costituita dall’area 4 e dall’area 6. È possibile distinguere l’area motoria primaria, deputata al controllo sia di singoli muscoli sia di piccoli gruppi muscolari, e l’area premotoria, che, sebbene la sua funzione non sia ancora completamente conosciuta, è possibile sia implicata nel fissare la postura all’inizio di un movimento pianificato e nel rendere l’individuo pronto per la prestazione. La maggior parte dei segnali, che da essa originano, provoca movimenti muscolari di una certa complessità che coinvolgono gruppi di muscoli per l’esecuzione di compiti specifici.
L’area motoria supplementare sembra essere coinvolta principalmente nella programmazione di sequenze motorie. Benché le aree 4 e 6 costituiscano la corteccia motrice, è importante riconoscere che il controllo corticale del movimento volontario coinvolge quasi tutta la neocorteccia [70] (Figura 2).

Figura 2. Suddivisione funzionale delle zone della corteccia cerebrale secondo la classificazione di Brodmann.
L’immagine mentale del corpo in movimento dipende dagli input visivi, propriocettivi e somatosensoriali che arrivano alla corteccia parietale posteriore. Vi sono due aree di particolare interesse nella corteccia parietale posteriore: l’area 5, che è il bersaglio degli input provenienti dalle aree corticali somatosensoriali 1, 2 e 3, e l’area 7 che è il bersaglio delle aree visive corticali di ordine superiore. Le aree prefrontali, insieme con la corteccia parietale posteriore, rappresentano il livello più alto della gerarchia del controllo motorio [71].
Il controllo motorio dunque è caratterizzato da due sistemi: pianificazione del movimento e controllo dell’esecuzione del movimento (Figura 3).
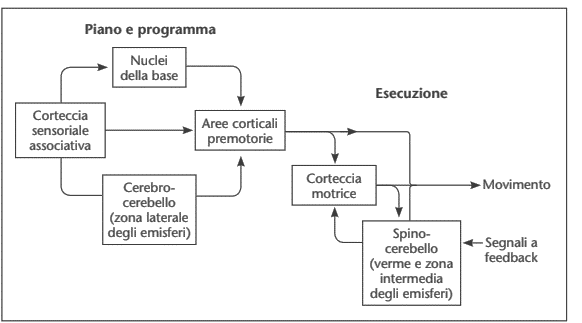
Figura 3. Sistemi di pianificazione ed esecuzione del movimento.
In sintesi, l’esecuzione del movimento prevede il coinvolgimento di: 1) aree corticali sensoriali; 2) aree motorie (pianificazione del movimento);3) cervelletto e gangli della base (confronto tra movimento pianificato e movimento eseguito; feed-forward; pianificazione).
Dal punto di vista delle funzioni, invece, le aree di sviluppo coinvolte nelle prassie sono rappresentate dalla:
Un disturbo della funzione motoria può dunque coinvolgere i diversi livelli tra cui la pianificazione, il controllo e l’esecuzione degli atti motori finalizzati e può interferire a sua volta con l’esecuzione dei diversi tipi di prassie: ideative, ossia azioni senza oggetto; ideomotorie, riferite ad azioni in presenza di uno o più oggetti; grafo-motorie, con problem solving spaziale; visuo-costruttive; oro-buccali-linguali; di sguardo; verbali [70].
Lo shifting è definibile come la capacità di adattarsi velocemente a una nuova situazione misurando le reazioni a stimoli che cambiano in modo costante. Tale abilità di adattamento permette all’individuo di essere autonomo e di agire attraverso un comportamento indipendente e intenzionale. La capacità di mutare strategie di pensiero e di azione per percepire ed elaborare informazioni e per far fronte alle situazioni si sviluppa tra i 7 e i 9 anni d’età per poi maturare intorno ai 12 anni.
L’abilità di «shiftare», ovvero di passare da un set all’altro di risposte risulta avere un ruolo predominante durante il processo di risoluzione di un problema. Tale meccanismo si compone di fasi diverse che prevedono: 1) la scomposizione del piano d’azione in vari step come la definizione e la rappresentazione del problema, 2) la formulazione di una strategia, 3) l’organizzazione delle informazioni, 4) il controllo del processo di soluzione e 5) la valutazione dell’efficacia della soluzione stessa [72].
I bambini che hanno difficoltà a «shiftare» da un compito all’altro non sono in grado di mutare il proprio comportamento in relazione al contesto e pertanto, di fronte alla risoluzione di un problema, compiono errori di perseverazione: danno una risposta scorretta e continuano a sbagliare, ignorando il feedback esterno [73].
L’inibizione è una componente dell’intelligenza che permette di sospendere le azioni e le decisioni per un tempo sufficiente a eseguire analisi cognitive più sofisticate e complesse, che consentono quindi un migliore adattamento ai cambiamenti ambientali.
Il concetto d’inibizione s’inserisce all’interno dei meccanismi dell’attenzione selettiva ed opera sia sull’informazione rilevante sia su quella non rilevante. La sua funzione, dunque, non è solo quella di facilitare l’elaborazione dell’informazione a cui si presta attenzione attraverso un meccanismo eccitatorio, ma anche quella di inibire, in maniera attiva e selettiva, l’elaborazione dell’informazione non rilevante meccanismo inibitorio [74]. Quest’ultimo meccanismo sopprime il processamento dell’informazione non rilevante, facendo in modo che i codici riguardanti quell’informazione siano meno disponibili ai meccanismi di risposta [75].
Il meccanismo di inibizione, dunque, svolge un ruolo fondamentale in vari processi cognitivi come memoria, attenzione selettiva e linguaggio [76]. Il periodo critico per lo sviluppo di tale abilità è intorno ai 9 anni [77].
L’ inibizione può essere di quattro tipi:
L’integrazione sensoriale è la capacità di ricevere, interpretare, scegliere e integrare le informazioni sensoriali che derivano dei recettori del tatto, della gravità e del movimento (vestibolari), e da quelli uditivi, visivi, gustativi e olfattivi, così come di rispondere in modo appropriato all’ambiente. La capacità di modulare o regolare i livelli degli input è necessaria per evitare l’ipo o l’iper stimolazione [79].
Il linguaggio di base si articola nei seguenti livelli di base: 1) fonemi, 2) morfemi, 3) sintassi, 4) semantica, 5) pragmatica, 6) discorso e 7) prosodia.
La comunicazione non verbale invece, deputata anch’essa a veicolare informazioni significative, opera attraverso una gamma di quattro o più canali: 1) il canale uditivo, 2) il canale visivo, 3) il canale tattile e 4) il canale cinestesico. Essa inoltre utilizza una varietà di codici come 1) i gesti, 2) il linguaggio del corpo, 3) le espressioni facciali, 4) lo sguardo, 5) la prosodia, 6) il contatto, 7) la prossemica e 8) la cronimica [81].
L’elaborazione dell’informazione uditiva è implicata anche nella comprensione del linguaggio. Essa consiste nelle seguenti fasi: analisi delle immagini sonore di parole o espressioni, appianamento dell’immagine sonora con il ricordo di parole o espressione e derivazione del significato delle parole o espressioni. Le lacune di questa capacità possono risultare in un disturbo dell’elaborazione uditiva centrale, una condizione in cui la comprensione del linguaggio parlato è compromessa [82].
L’elaborazione visuo-spaziale si riferisce a un insieme di funzioni mediate dall’emisfero destro, che includono il riconoscimento di volti, delle espressioni emotive, l’orientamento destra-sinistra, la rotazione mentale di figure e la comprensione dei segnali della comunicazione non verbale. La percezione visiva implica la memoria visiva sequenziale, la discriminazione figura-sfondo, la memoria visiva, la chiusura visiva e la costanza della forma visiva. L’elaborazione spaziale implica la comprensione delle dimensioni spaziali del proprio corpo e dell’ambiente fisico [83] [84].
“Chiunque sa cosa sia l’attenzione. È la mente che prende possesso, in forma chiara e vivida, di uno solo tra i vari oggetti o le varie catene di pensieri simultaneamente possibili” [85].
L’attenzione è un processo psicologico che permette alle persone di essere selettivamente consapevoli di una parte o di un aspetto dell’ambiente sensoriale e di rispondere in modo selettivo a una classe di stimoli [86].
L’ambiente che ci circonda invia infatti al nostro cervello una moltitudine d’informazioni che possono essere visive, acustiche, tattili e olfattive. Tuttavia, poiché il nostro sistema cognitivo ha un numero di risorse limitate, per evitare una situazione di «sovraccarico», solo una parte di tali informazioni in ingresso sono elaborate approfonditamente per poi diventare coscienti. Questa capacità di selezionare gli stimoli rilevanti all’interno del flusso di informazioni sempre disponibili è vitale perché consente di controllare l’ambiente e mettere in atto comportamenti adeguati alle situazioni.
L’elaborazione di uno stimolo prevede diversi stadi di processamento: gli studiosi sono ormai concordi nel descrivere un’iniziale 1) elaborazione delle caratteristiche fisiche (semplici e complesse) di uno stimolo, quali ad esempio il suo orientamento nello spazio, la forma, la dimensione, il colore; una successiva fase consiste 2) nell’elaborazione semantica, ossia nell’estrazione del suo significato; infine, avviene 3) la selezione della risposta appropriata per lo scopo prefissato in relazione a quel particolare stimolo [87].
Le prime teorie facevano riferimento all’attenzione come a una funzione di «filtro» che operando ai primi livelli, precoci, di elaborazione sensoriale dell’informazione, lasciava passare solo le informazioni rilevanti [88]. Le evidenze sperimentali hanno però disconfermato questo modello, ipotizzando una diversa posizione del filtro attentivo nell’elaborazione degli stimoli: infatti, in seguito a una prima fase in cui tutte le informazioni vengono analizzate, solo i segnali più rilevanti sono riconosciuti e selezionati; le altre informazioni restano disponibili ed eventualmente possono essere richiamate in altre situazioni [89].
Successive formulazioni teoriche, hanno abbandonato totalmente l’idea del filtro attentivo, riferendosi all’attenzione in termini di capacità e di quantità. Disporremmo, cioè, di una quantità fissa di attenzione, che verrebbe allocata secondo ciò che il compito richiede. L’attenzione agirebbe come un meccanismo deputato alla programmazione e al controllo dei processi cognitivi in relazione agli scopi del soggetto, con l’obiettivo di evitare che stimoli in conflitto tra loro ostacolino il funzionamento cognitivo [90].
Allo stato attuale l’interrogativo principale riguarda l’esistenza di un modello unico che coordini tutti i vari tipi di attenzione oppure di più modelli distinti. La neuropsicologia cognitiva moderna tende a descrivere il funzionamento cognitivo come un’organizzazione gerarchica, alla cui base ci sono le operazioni di routine, mentre ai vertici vi sarebbe una struttura coordinatrice di gestione e controllo [57].
L’attenzione è un insieme di sottocomponenti distinte ma interagenti, è un unico fenomeno caratterizzato da diversi aspetti ai quali facciamo riferimento.
L’arousal o allerta è lo stato fisiologico di attivazione dell’individuo; si può suddividere in un’allerta tonica intesa come la capacità di mantenere un adeguato livello di prestazione per un certo periodo e un’allerta fasica, cioè un incremento della capacità di risposta in seguito alla comparsa di un segnale di avvertimento [87].
L’attenzione selettiva è la capacità di selezionare una parte degli stimoli in entrata e sottoporli a un’elaborazione accurata, mentre i restanti vengono elaborati solo parzialmente e in modo più superficiale. Di solito, scegliamo di prestare maggiore attenzione alle informazioni che sono rilevanti per l’attività che intendiamo svolgere e che sono utili al raggiungimento degli scopi che ci siamo prefissi. La situazione che costituisce il classico esempio di attenzione selettiva è rappresentata dal cosiddetto effetto «cocktail party»: nonostante tutte le emissioni sonore provenienti dall’ambiente circostante siano colte dai nostri recettori acustici, siamo in grado di selezionare e analizzare solo quelle provenienti dalla persona con la quale stiamo conversando. La funzione dell’attenzione selettiva, infatti, consiste proprio nell’evitare interferenze tra stimoli compresenti in modo tale che le azioni siano coordinate e finalizzate all’obiettivo preposto [91].
L’attenzione divisa è la capacità di prestare attenzione a più stimoli simultaneamente. La situazione sperimentale tipica nello studio dell’attenzione divisa è il paradigma del doppio compito, che impegna i partecipanti contemporaneamente in due attività, che possono essere di difficoltà diversa o che impegnano abilità diverse: il risultato generalmente osservabile in questa situazione è che la prestazione nei due compiti cognitivi è migliore di quella rilevabile se i due compiti sono eseguiti simultaneamente.
Nell’ambito del paradigma del doppio compito, quando i due compiti necessitano entrambi di processi di controllo, si osserva un peggioramento delle prestazioni. Se uno dei due compiti richiede invece un processo automatico e l’altro un processo di controllo, difficilmente si avrà un’interferenza tra le due prestazioni.
L’attenzione sostenuta è la capacità di prestare attenzione a un ben preciso campo di stimolazione per un periodo prolungato. Tale componente attentiva permette di mantenere un’adeguata prestazione in compiti monotoni per periodi relativamente lunghi.
Diversi fattori possono influenzare la prestazione in compiti di attenzione sostenuta: ad esempio l’uso di segnali degradati, la cui ampiezza e durata sono appena al di sopra della soglia percettiva, un ritmo di presentazione degli stimoli non costante, oppure l’incertezza spaziale o temporale degli stimoli che può determinare un peggioramento della performance [87]. Cosa realmente distingue l'attenzione sostenuta dalle altre componenti attentive è da ricercare nelle prestazioni su una singola attività nel corso del tempo e nelle fluttuazioni all'interno di un individuo della capacità complessiva di mantenere la prestazione di un compito stabile.
Il modo più semplice per misurare l'attenzione è quello di misurare le fluttuazioni di attenzione prolungata su una prestazione momento per momento. Esaminare in questo modo il processo di attenzione ha permesso ai ricercatori di tracciare fluttuazioni in prestazioni con un alto grado di risoluzione temporale. Di conseguenza, è stato recentemente introdotto un metodo per esplorare fluttuazioni intraindividuali a tempo di reazione, chiamato “decorso di varianza temporale” (DVI).
L'analisi del DVI ha rivelato fluttuazioni dell’attenzione tra periodi di bassa variabilità, alta precisione, piccoli aggiustamenti di errori correlati nella zona dello stato attentivo e tra periodi di variabilità superiore, bassa precisione, e aggiustamenti più grandi di errori correlati (fuori dalla zona dello stato attentivo), attraverso molteplici attività. Queste fluttuazioni, nella loro variabilità, in realtà sarebbero ampiamente indipendenti dai decrementi di vigilanza.
Di seguito descriveremo i modelli neurocognitivi che si prefiggono di spiegare i fattori chiave che fanno riferimento all’attenzione sostenuta (Figura 4).
![Figura 4. Le prestazioni di un compito sono modulate dall’Arousal, dall’allocazione attentiva (Attentional Allocation) e dall’elaborazione delle informazioni (Information processing). L’Arousal consentirà un livello sufficiente di risorse cognitive / attenzionali (in giallo) e il controllo cognitivo determinerà la proporzione delle risorse disponibili che saranno dedicate all'attività, a loro volta influenzate dal costo intrinseco di controllo (Intrinsic cost) e dalla motivazione (Motivation). In un basso stato di Arousal ci saranno meno risorse cognitive da allocare e quindi l'esecuzione delle attività potrebbe non essere ottimale, anche se quelle risorse saranno principalmente dedicate all'attività. In uno stato di grande eccitazione / distrazione, potrebbero esserci risorse sufficienti ma l'attenzione sarà meno selettiva, così come sarà diretta anche verso processi mentali non correlati al compito [92].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/figura-4-arousal-attentiva-attentional-allocation-elaborazione-informazioni-information-processing.png)
Figura 4. Le prestazioni di un compito sono modulate dall’Arousal, dall’allocazione attentiva (Attentional Allocation) e dall’elaborazione delle informazioni (Information processing). L’Arousal consentirà un livello sufficiente di risorse cognitive / attenzionali (in giallo) e il controllo cognitivo determinerà la proporzione delle risorse disponibili che saranno dedicate all'attività, a loro volta influenzate dal costo intrinseco di controllo (Intrinsic cost) e dalla motivazione (Motivation). In un basso stato di Arousal ci saranno meno risorse cognitive da allocare e quindi l'esecuzione delle attività potrebbe non essere ottimale, anche se quelle risorse saranno principalmente dedicate all'attività. In uno stato di grande eccitazione / distrazione, potrebbero esserci risorse sufficienti ma l'attenzione sarà meno selettiva, così come sarà diretta anche verso processi mentali non correlati al compito [92].
La memoria è quella funzione cognitiva che permette l’acquisizione, l’immagazzinamento e il recupero di informazioni provenienti da diversi input sensoriali.
Le abilità mnesiche sono organizzate in diversi moduli funzionalmente e strutturalmente autonomi, seppure interagenti tra loro. Si distingue, infatti, in due magazzini, uno a breve termine (MBT) e uno a lungo termine (MLT). [93] Tali sistemi si differenziano per capacità di ritenzione: limitata a pochi elementi per la MBT e praticamente illimitata per la MLT; la codifica dell’informazione è di tipo prevalentemente fonologico nel primo caso, basata su un’elaborazione semantica nel secondo; la velocità di decadimento della traccia mnesica è limitata a pochi secondi in assenza di reiterazione per la MBT, variabile ma comunque lenta per la MLT [94]. Un importante contributo alla descrizione del funzionamento della memoria è costituito dal modello cognitivo della Memoria di Lavoro (ML) tra cui quello di maggiore rilevanza è il Modello di Baddeley e Hitch del 1974, successivamente perfezionato dallo stesso Baddeley nel 1986:
La ML è un sistema mnesico a capacità limitata che immagazzina temporaneamente le informazioni per una successiva manipolazione. Non si tratta di un magazzino unitario, ma si articola in diverse sottocomponenti [57] (Figura 5).

Figura 5. Modello cognitivo della ML secondo Baddeley e Hitch (1974).
Il primo sistema, detto sistema esecutivo centrale, è una sorta di elaboratore centrale a capacità limitata, in grado di processare informazioni appartenenti a più modalità sensoriali. È un sistema flessibile responsabile anche del controllo e della regolazione dei processi cognitivi. L’esecutivo centrale è supportato da una serie di sistemi schiavi in grado di ritenere temporaneamente informazioni appartenenti a una sola modalità sensoriale.
Il più studiato è il loop articolatorio, un sistema specializzato per la ritenzione temporanea di materiale verbale composto da un magazzino fonologico per la ritenzione dell’informazione verbale e da un meccanismo di ripasso subvocalico («refreshment»), con il compito di evitare il decadimento delle informazioni [95].
Il secondo sistema è il taccuino visuo-spaziale, specializzato nel processamento e nel mantenimento di materiale visivo e spaziale.
Nel modello originario non è prevista alcuna modalità di interazione tra il loop articolatorio e il taccuino visuo-spaziale, né tantomeno un ruolo della memoria sulla conoscenza cosciente, che invece dovrebbe essere di cruciale importanza [96]. Pertanto, lo stesso Baddeley ha successivamente perfezionato il modello originario, ipotizzando il coinvolgimento di un buffer episodico, che opera sotto il controllo dell’esecutivo centrale con il compito di ritenere e integrare le informazioni provenienti dagli altri due sistemi schiavi così da formare episodi integrati e coscienti [97] [98] (Figura 6).

Figura 6. Modello cognitivo della ML di Baddeley (2000).
Ad oggi con il termine “architettura cognitiva” si intende riferirsi ad insiemi di regioni cerebrali che contribuiscono alla performance di di attività correlate, o di un particolare insieme di funzioni. Spesso queste architetture sono esplicitamente indicate come reti (networks): ad esempio il network con modalità di default ed i network dell’attenzione. Tuttavia, il significato del termine “network” è molto variabile. In molti casi, l’utilizzo informale di questo termine viene applicato ad una semplice raccolta di regioni che si attivano durante una serie di relativi studi di imaging fMRI, senza alcun riferimento esplicito alle connessioni tra queste regioni. In contrasto con questo concetto informale di reti come insiemi di regioni vi è la definizione più formale di ciò che costituisce un network: esso è un insieme di relazioni a coppie tra gli elementi di un sistema formalmente rappresentato come un insieme di archi che collegano un insieme di nodi. I network neurobiologici giungono a più livelli di una scala a partire da percorsi metabolici e regolatori cellulo-specifici all'interno di neuroni che interagiscono con i sistemi di aree corticali e nuclei sub corticali [99] (Figura 7).
![Figura 7. Rappresentazione schematica dei livelli di struttura all'interno del sistema nervoso [100].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/figura-7-rappresentazione-schematica-livelli-struttura-interno-sistema-nervoso.png)
Figura 7. Rappresentazione schematica dei livelli di struttura all'interno del sistema nervoso [100].
Ad ogni livello (neuroni, circuiti neuronali, sistemi e popolazioni) sono presenti diversi tipi di reti con differenti proprietà. A ciascuno di questi livelli, è importante non solo capire come funzionano i singoli elementi, ma soffermarsi anche sulle strutture di relazioni a coppie che mettono gli elementi nel contesto del sistema interconnesso più grande [101]. Con alcune eccezioni, le architetture cognitive principalmente coinvolgono strutture e meccanismi a questo livello massimo di analisi [102]. Alla base della regolazione della maggior parte, se non di tutti i tratti comportamentali e fisiologici, tra cui l'eccitazione, il sonno, la motivazione, l'emozione e la memoria, vi sono neuromodulatori come l’acetilcolina, le monoamine e i neuropeptidi [103]. Una caratteristica peculiare di questi sistemi è che contengono più di un singolo trasmettitore o modulatore: la neuromodulazione, ad esempio di ammine e neuropeptidi, si verifica contemporaneamente alla neurotrasmissione classica di neurotrasmettitori a piccole molecole, che si pensa offra flessibilità alla rete neurale e maggiore complessità nell’integrazione sinaptica delle informazioni trasmesse [104].
I neurotrasmettitori sono sostanze chimiche sintetizzate e immagazzinate nei neuroni, liberate nella chiave sinaptica durante l’attività nervosa, che interagiscono con i recettori specifici della membrana postsinaptica e pertanto sono responsabili di un cambiamento nell’attività delle cellule postsinaptiche che può essere di tipo inibitorio o eccitatorio [105].
È necessario aggiungere che un certo numero di sostanze tradizionalmente classificate come neurotrasmettitori sono anche neuromodulatori o svolgono una funzione neuromodulatrice: i neuromodulatori non modificano direttamente il potenziale di membrana neuronale come i neurotrasmettitori, ma influenzano l’efficienza del processo di neurotrasmissione [106].
Il prosencefalo basale (BF) dà origine a uno dei centri neuromodulatori meglio caratterizzati, il sistema di proiezione colinergica. Gli alberi insolitamente lunghi degli assoni dei neuroni colinergici innervano praticamente tutte le aree corticali e molti centri sottocorticali (Figura 8).
![Figura 8. Centri neurali a doppio neurotrasmettitore che regolano l'arousal, l'attenzione, l'apprendimento e la memoria. (A) Proiezioni colinergiche del prosencefalo basale (BF). I siti noti di co-trasmissione sono contrassegnati dal doppio colore. (B) Proiezioni istaminergiche dal nucleo tuberomammillare (TMN). (C) Proiezioni dell’orexina dall’ipotalamo laterale (LH). (D) Proiezioni di relaxina-3 dal nucleo incertus (NI). Le interazioni note tra questi sistemi sono contrassegnate da contorni tratteggiati [107].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/figura-8-centri-neurali-doppio-neurotrasmettitore-regolano-arousal-attenzione-apprendimento-memoria.png)
Figura 8. Centri neurali a doppio neurotrasmettitore che regolano l'arousal, l'attenzione, l'apprendimento e la memoria. (A) Proiezioni colinergiche del prosencefalo basale (BF). I siti noti di co-trasmissione sono contrassegnati dal doppio colore. (B) Proiezioni istaminergiche dal nucleo tuberomammillare (TMN). (C) Proiezioni dell’orexina dall’ipotalamo laterale (LH). (D) Proiezioni di relaxina-3 dal nucleo incertus (NI). Le interazioni note tra questi sistemi sono contrassegnate da contorni tratteggiati [107].
Recenti studi hanno dimostrato che, oltre all’acetilcolina (ACH), i neuroni colinergici del prosencefalo possono rilasciare altri neurotrasmettitori. Si è osservato che due sistemi colinergici separati, gli interneuroni tonicamente attivi dello striato e le cellule colinergiche dell’habenula mediale, possono rilasciare sia ACH che glutammato [103].
Un recente studio ha inoltre dimostrato che le cellule colinergiche possono rilasciare anche il trasmettitore inibitorio GABA [108] e che la co-trasmissione di ACH e GABA potrebbe controllare la plasticità e l'apprendimento corticale. A livello di circuito, è stato dimostrato infatti che ACH è in grado di desincronizzare reti corticali, diminuire correlazioni tra coppie di neuroni e aumentare l'attivazione corticale in generale. È importante inoltre sottolineare che ACH ha una forte influenza sulla plasticità sinaptica, sia in termini di controllo di campi recettivi sensoriali che di curve di ottimizzazione. A livello comportamentale, la capacità dei neuroni colinergici centrali di aumentare l'attivazione corticale e ottimizzare la plasticità sinaptica può essere alla base dei loro effetti ampiamente osservati sull'attenzione, sull'apprendimento e sulla memoria. [103] Comprendere le contingenze comportamentali che determinano i tempi precisi di rilascio dell’acetilcolina informerebbe su come i neuroni colinergici mediano diversi processi cognitivi: misurando il rilascio di ACH con sonde voltmetriche sensibili alla colina, è possibile osservare che i transitori colinergici nella corteccia prefrontale mediale sono correlati al rilevamento di segnali visivi in un compito di attenzione prolungato. Ma questi transitori colinergici sono risultati assenti dopo rilevamenti consecutivi e quindi sono stati associati a degli spostamenti di attenzione.
Un ulteriore e recente studio ha sondato ed identificato l’attività dei neuroni colinergici del nucleo basale dei topi e del nucleo orizzontale della banda diagonale di Broca, due parti distinte del sistema colinergico del BF. I topi sono stati addestrati in un compito di rilevamento uditivo che richiedeva attenzione sostenuta e l'apprendimento di un rinforzo incorporato. Sorprendentemente, mentre alcuni neuroni colinergici non presentavano forti correlazioni attentive, neuroni colinergici del BF sono stati invece attivati con breve latenza e alta precisione a seguito di una punizione e di un premio. Le risposte colinergiche dunque sono state ridimensionate con l'imprevisto del rinforzo, cioè con un "rinforzo a sorpresa", o più semplicemente, i neuroni colinergici hanno risposto quando la ricompensa era inaspettata, mostrando invece una risposta scarsa o nulla alla ricompensa attesa [109].Questi risultati suggeriscono come i neuroni colinergici possano essere fortemente coinvolti nel processo di apprendimento attraverso il rinforzo, e come siano correlati a un’attivazione corticale precisa rispetto ad una sua consegna.
Altri recenti studi dimostrano come:
Le bambine con RTT dopo un breve periodo di sviluppo apparentemente quasi normale regrediscono con perdita del linguaggio acquisito e dell’uso funzionale delle mani, sostituito da stereotipie caratteristiche della mano e con un deterioramento cognitivo profondo. Esse presentano infatti un funzionamento cognitivo profondamente alterato; stime tipiche hanno suggerito uno sviluppo mentale compreso tra gli 8 e i 12 mesi [110].Tuttavia, numerose osservazioni e rapporti dei genitori indicano che molti individui con RTT sono in grado di stabilire un contatto visivo tale da poter indicare e comunicare attraverso lo sguardo, suggerendo che essi possano avere capacità cognitive più evidenti di quanto si pensi [111].La valutazione cognitiva è senza dubbio una sfida in individui con disabilità fisiche profonde, in particolare in quelli in cui è carente anche l’uso funzionale delle mani e del linguaggio. Di conseguenza, i metodi di prova convenzionali, che richiedono risposte di tipo motorio o verbale o entrambi, sono inadeguate per i bambini con RTT.I recenti progressi tecnologici nell’ambito del contatto oculare (eye-tracking) tuttavia, offrono un modo per le persone con RTT di compiere scelte e di comunicare in modo indipendente [110]. In alcuni studi iniziali di Djukic e colleghi si è potuto osservare che i bambini con questa patologia sono in grado di esplorare nuovi stimoli con i loro occhi in modo simile a bambini neurotipici, suggerendo che possano elaborare il significato degli stimoli in modo paragonabile ai loro coetanei [112].In un nuovo documento del Journal Pediatric Neurology, Ahonniska-Assa e colleghi hanno voluto esaminare le capacità cognitive di ragazze con RTT più direttamente modificando un test standard del vocabolario ricettivo, il Peabody Picture Vocabulary Test, in un gruppo di 17 ragazze con RTT. Anche se questo test non richiede una risposta verbale, i partecipanti sono tenuti ad indicare l’immagine tra le quattro che è associata ad un prompt suggerito dal somministratore del test. Visualizzate le quattro immagini poste sul monitor di un computer, i soggetti poi dovevano indicare la loro risposta cercando in una delle immagini. La risposta veniva registrata dal sistema di un puntatore oculare. Lo studio ha rilevato che quasi un terzo delle ragazze con RTT che avevano effettuato il test, ha mostrato un vocabolario ricettivo con un livello indicativo di lieve deficit cognitivo (2/17) o persino all'interno dell'intervallo normale (4/17) [113]. Questo è un risultato importante che ha forti implicazioni per la nostra comprensione delle capacità cognitive nella RTT.Questo test potrebbe aver sopravvalutato le capacità di RTT?Dato il protocollo del test, questo sembra improbabile. Non è da escludere invece la possibilità che i risultati abbiano sottostimato il potenziale delle ragazze con RTT. Lo sguardo non è una tecnica facile da padroneggiare. La maggior parte delle ragazze non era abituata ad utilizzare i dispositivi per lo sguardo all'inizio dello studio e i dati normativi riguardano soggetti in grado di indicare. Inoltre, l'esposizione al linguaggio, e sicuramente alcuni input educativi, possono benissimo differire nei bambini dove si presume ci sia una grave compromissione cognitiva [110].In un ulteriore recente studio è stata utilizzata la tecnologia dell’eye-tracking per valutare i punti di forza e i limiti cognitivi associati con RTT. Inizialmente è stata esaminata la possibilità d'utilizzo di questa nuova tecnica, con cui si dimostrerebbe che gli individui con RTT preferiscono stimoli socialmente ponderati, si concentrano selettivamente su aree salienti e nuovi elementi di una scena e sono in grado di tracciare tratti sia orizzontali che verticali. Altri studi sulla memoria di riconoscimento invece, condurrebbero a constatare che sebbene le ragazze con RTT siano in grado di riconoscere schemi semplici, volti e alcune espressioni emotive, le loro prestazioni risulterebbero essere significativamente più scarse di quelle di bambini con uno sviluppo tipico [111].
Sono state riportate anche atipicità nell'attenzione, compresa una ricerca mal distribuita e meno attenta delle aree chiave durante l’esecuzione di un compito, associata ad una memoria più povera. Tale deficit di attenzione è risultato rilevante dato il ruolo che questa ha in molte aree, tra cui la memoria di lavoro e facoltà cognitive più complesse, compresi il linguaggio, e le funzioni esecutive.L’attenzione era inizialmente considerata come un sistema unitario, mentre ora è riconosciuto come un sistema composto da diversi processi correlati ma separabili, sottoposti a distinti sistemi neurali. Due dei processi fondamentali riguardano lo spostamento e il mantenimento dell'attenzione. Lo spostamento e il mantenimento dell'attenzione da uno stimolo ad un altro può essere guidata da fattori esogeni di basso livello, oppure da fattori endogeni di ordine superiore. Quando l’attenzione è sotto controllo esogeno, gli spostamenti sono reattivi, automatici o governati da cambiamenti esterni al campo visivo; quando è sotto controllo endogeno gli spostamenti dipendono in gran parte dalle intenzioni del soggetto e si basano su reti neurali frontali coinvolte nell'attenzione esecutiva: un esempio tipico di spostamento endogeno è una saccade anticipatoria, quando viene generato un movimento oculare verso uno stimolo che ancora non è presente.Il paradigma che sposta lo sguardo ha permesso di valutare sia l'attenzione guidata endogena (anticipazione dell'aspetto di un bersaglio) sia che quella guidata esogena (risposta all'apparizione di un bersaglio), mentre contemporaneamente è stata valutata l'attenzione sostenuta. In particolare, lo studio ha comportato una serie di prove in cui appariva un attrattore centrale, insieme a due finestre vuote, una per lato; dopo un tempo di 1000 ms, appariva un target nella finestra a destra. Lo spostamento di attenzione è stato misurato dalle saccadi reattive e anticipatorie sulla posizione target. L'attenzione sostenuta è stata misurata dal tempo trascorso a guardare il bersaglio.Il campione finale era composto da 20 femmine con diagnosi clinica di RTT classica. Il gruppo di confronto era composto da 14 bambine con sviluppo tipico (che chiameremo gruppo ST). Dal gruppo ST sono stati esclusi bambini con disturbi neurologici significativi (ad es. epilessia, tumore al cervello), compromissioni sensoriali, disturbi dello sviluppo neurologico (ad es. autismo, disturbo da deficit di attenzione e iperattività), o con parenti di primo grado con disturbi dello sviluppo neurologico. Il tempo impiegato dai bambini a spostare lo sguardo dall'attrattore alla posizione target è mostrato nel grafco 3.

Grafico 3. Tempo impiegato per spostare l'attenzione dall'attrattore centrale al target nei blocchi di prove.
Il gruppo RTT aveva tempi di reazione costantemente più lunghi in tutti i sei blocchi di prove in cui ha impiegato, in media, 504 ms di tempo in più per rivolgersi al target rispetto al gruppo ST. Inoltre, poiché il tempo impiegato per spostare l’attenzione verso l'obiettivo avrebbe potuto comportare movimenti oculari saccadici anticipatori o reattivi, sono stati esaminati separatamente anche questi due elementi.· movimenti saccadici anticipatori: poiché la posizione del target imminente era prevedibile, alcuni dei movimenti oculari dei bambini avrebbero potuto essere naturalmente anticipati, ovvero nell'intervallo di 1000 ms dopo la comparsa dell'attrattore centrale e prima della comparsa del target. La figura 13 indica che i bambini ST hanno mostrato anticipazioni di questi movimenti su quasi il 60% delle prove, mentre i bambini con RTT lo hanno fatto in meno del 20% delle prove;· movimenti saccadici reattivi: questi movimenti riflettono la rapidità con cui il bambino sposta l'attenzione al bersaglio dopo che è apparso. Anche se i bambini del gruppo ST hanno reagito un più rapidamente rispetto ai bambini con RTT di circa 100 ms, questa differenza invece non era significativa (Grafico 4).

Grafico 4. (A) Media percentuale di anticipazioni per bambino in media in ogni gruppo. (B) Tempo medio di movimenti saccadici reattivi per bambino in ogni gruppo.
Queste scoperte indicano che i bambini con RTT potrebbero mantenere l'attenzione su uno stimolo attraverso il loro sguardo tipico che viene spesso individuato come una caratteristica della malattia, e orientarlo abbastanza rapidamente quando appare un bersaglio ben visibile. Tuttavia, risultano avere una grande difficoltà nell'anticipare eventi prevedibili e nell’utilizzare un’attenzione di tipo endogeno che ha implicazioni deleterie per il funzionamento esecutivo.La scarsità di anticipazione nei movimenti saccadici nei bambini con RTT suggerisce che vi è una difficoltà a (1) formare una rappresentazione interna della regola che governa l'aspetto del target o (2) ad agire sulle rappresentazioni interne che sono state formate. La difficoltà di formare o utilizzare una tale rappresentazione interna, condizione sine qua non per le funzioni esecutive, li porrebbe in condizioni di svantaggio nella pianificazione delle risposte ambientali senza la possibile determinazione di obiettivi interni e nella gestione delle situazioni sociali.L'attenzione esecutiva è associata alla corteccia prefrontale cingolata anteriore, laterale e ventrale e ai gangli della base. La rete fronto-parietale coinvolta nell'attenzione esecutiva mostra una maggiore connettività funzionale durante lo sviluppo. L'assenza di miglioramenti legati all'età nelle prestazioni nel gruppo RTT aumenta la possibilità che vi siano connessioni e attivazioni frontoparietali ancora immature. Il presente studio dunque dimostra che spostamenti di attenzione guidati endogeni (anticipazioni), che coinvolgono l'attenzione esecutiva in soggetti con RTT, risultano essere gravemente compromessi, mentre spostamenti di attenzione esogeni, ovvero reazioni alla comparsa di un bersaglio altamente coinvolgente nel campo visivo, risultano essere relativamente intatti. L’attenzione sostenuta, sebbene più povera di quella dei bambini ST, era comunque considerevole, nei pazienti con RTT vicina al 75% del tempo disponibile speso guardando l'obiettivo. Potrebbe essere quindi possibile per migliorare la capacità dei bambini con RTT, anticipare eventi imminenti e accorciare l'intervallo tra un attrattore e un target [111].Se i pazienti con RTT presentano un’alterazione lieve del funzionamento cognitivo, o che rientra nel normale intervallo di intelligenza, devono essere in ogni caso adottate strategie educative e supporti comunicativi quotidiani adeguati. La comunicazione attraverso il puntatore oculare e il controllo di sguardo può essere la chiave per sbloccare il potenziale delle persone con RTT [110].
Uno studio che prenderemo in esame ha cercato di utilizzare i punti di forza visivi di cui sarebbero dotati i soggetti con RTT, aggirando le disabilità motorie e verbali associate con la sindrome, per la valutazione dell'attenzione e della memoria di riconoscimento utilizzando il paradigma del confronto visivo (che chiameremo PCV) in correlazione con la tecnologia eye-tracking.Nel paradigma PVC, due stimoli identici vengono brevemente presentati per la familiarizzazione e successivamente lo stimolo familiare viene messo a confronto con uno nuovo durante il test; il riconoscimento è dedotto dallo sguardo preferenziale diretto al nuovo target [114] [115].Si presume che nel corso della prova venga creata una rappresentazione mentale del bersaglio durante la familiarizzazione e, che quando si incontra un nuovo target nel test che non corrisponde alla rappresentazione, l’attenzione si sposti su quello nuovo.Questa coorte di studio comprendeva 27 bambine (età media 10 AA) con RTT classica diagnosticata clinicamente e 30 con sviluppo normale (età media 10 AA). [116]La batteria PVC utilizzata consisteva di 9 problemi: cinque con foto acromatiche di volti e quattro con motivi astratti multicolori [117] (Figura 9).
![Figura 9. Gli stimoli per l'attività PVC [114].](https://www.neuropsicomotricista.it/images/stories/tesi-di-laurea/francesca-guerri/figura-9-gli-stimoli-per-attivita-pvc.png)
Figura 9. Gli stimoli per l'attività PVC [114].
Per ridurre al minimo la misura in cui la variazione procedurale potrebbe contribuire alle differenze tra i partecipanti, i problemi sono stati somministrati in un ordine fisso, cominciando con le facce della coppia in alto e quindi procedendo verso il basso, intervallando gli stimoli del viso con gli altri modelli; i nuovi target e quelli familiari sono stati mantenuti costanti per ogni stimolo per tutti i partecipanti, con la figura sinistra di ogni coppia come target familiare e quella giusta come nuovo target; gli stimoli utilizzati sono stati pretestati per garantire che i membri di ciascuna coppia fossero approssimativamente uguali nell'attrattiva, così da minimizzare ogni possibile distorsione dovuta alle preferenze di stimolo.
Il riconoscimento viene quantificato in base ad un di punteggio novità ovvero alla percentuale di sguardi rivolti al nuovo target durante la prova.L'attenzione è stata valutata mediante misure del tempo di visione totale, del numero di fissazioni e della durata media delle fissazioni; la memoria di riconoscimento è stata valutata attraverso i punteggi novità.Nel complesso, sia per il gruppo RTT che il gruppo con sviluppo normotipico, è risultata una preferenza per gli occhi ma il gruppo RTT ha mostrato meno interesse generale per tutte le caratteristiche rispetto al gruppo di confronto.Il 44% delle bambine (n = 11) del gruppo RTT ha dimostrato assenza di interesse per il naso o per la bocca ignorandoli entrambi e non riuscendo nemmeno a guardare la bocca, mentre nel gruppo di confronto nessuno mostrava così poca attenzione a qualsiasi stimolo.Ciononostante, entrambi i gruppi sono stati in grado di discriminare i target associati sia all'uno che all'altro, e a riconoscere quello familiare.Punteggi novità più elevati sono stati correlati in entrambi i gruppi a diverse variabili di familiarizzazione quali la considerazione maggiore per l’aspetto più generale, maggiori fissazioni, maggiore percentuale di tempo impiegato per guardare lo stimolo e maggiori fissazioni agli occhi dei volti. I punteggi novità sono stati significativamente più alti per il gruppo di confronto. Sebbene questa differenza tra i sottogruppi sia stata significativa, i punteggi novità dei pazienti con RTT che osservavano più ampiamente erano di gran lunga inferiori a quelli dei partecipanti con sviluppo tipico.I risultati hanno indicato dunque che i pazienti con RTT potrebbero riconoscere volti e schemi sebbene, rispetto al confronto con l'età dei partecipanti, il loro riconoscimento sia stato più scarso. È da notare infatti che, nonostante le marcate differenze tra i motivi a colori e il design, che avrebbero dovuto facilitare il riconoscimento, il gruppo RTT aveva ancora più difficoltà a ricordarle di quanto non facessero gli altri partecipanti. L’utilizzo dell’eye tracking ha rivelato che il gruppo RTT ha mostrato un notevole interesse per i volti, dedicando circa il 70% del tempo di familiarizzazione disponibile per guardarli. Essi mostravano anche una decisa preferenza per gli occhi (rispetto al naso e alla bocca) - una preferenza forte come quella vista nell’altro gruppo [116]. Questa attrazione per il sociale, agli stimoli e l'attenzione per gli occhi si mostra in contrasto con ciò che succede nell’ autismo, dove l’interesse per i volti è ridotto e spesso il contatto oculare viene evitato [118].Nel complesso, gli sguardi caratterizzati da una quantità più elevata di fissazioni e da una minore lunghezza delle stesse, erano associati a un migliore riconoscimento facciale.Questi risultati indicano che la tecnologia eye-tracking, congiuntamente a compiti non verbali, ha il potenziale per sbloccare il mondo cognitivo di persone con RTT.
| Indice |
| INTRODUZIONE |
| CONCLUSIONI |
| BIBLIOGRAFIA |
| Tesi di Laurea di: Francesca GUERRI |
























